
Description of the Three Complete Mitochondrial Genomes of Sitta (S. himalayensis, S. nagaensis, and S. yunnanensis) and Phylogenetic Relationship (Aves: Sittidae)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and DNA Extraction
2.2. Genome Sequencing, Assembly, and Annotation
2.3. Phylogenetic Analyses
3. Results
3.1. Genome Organization
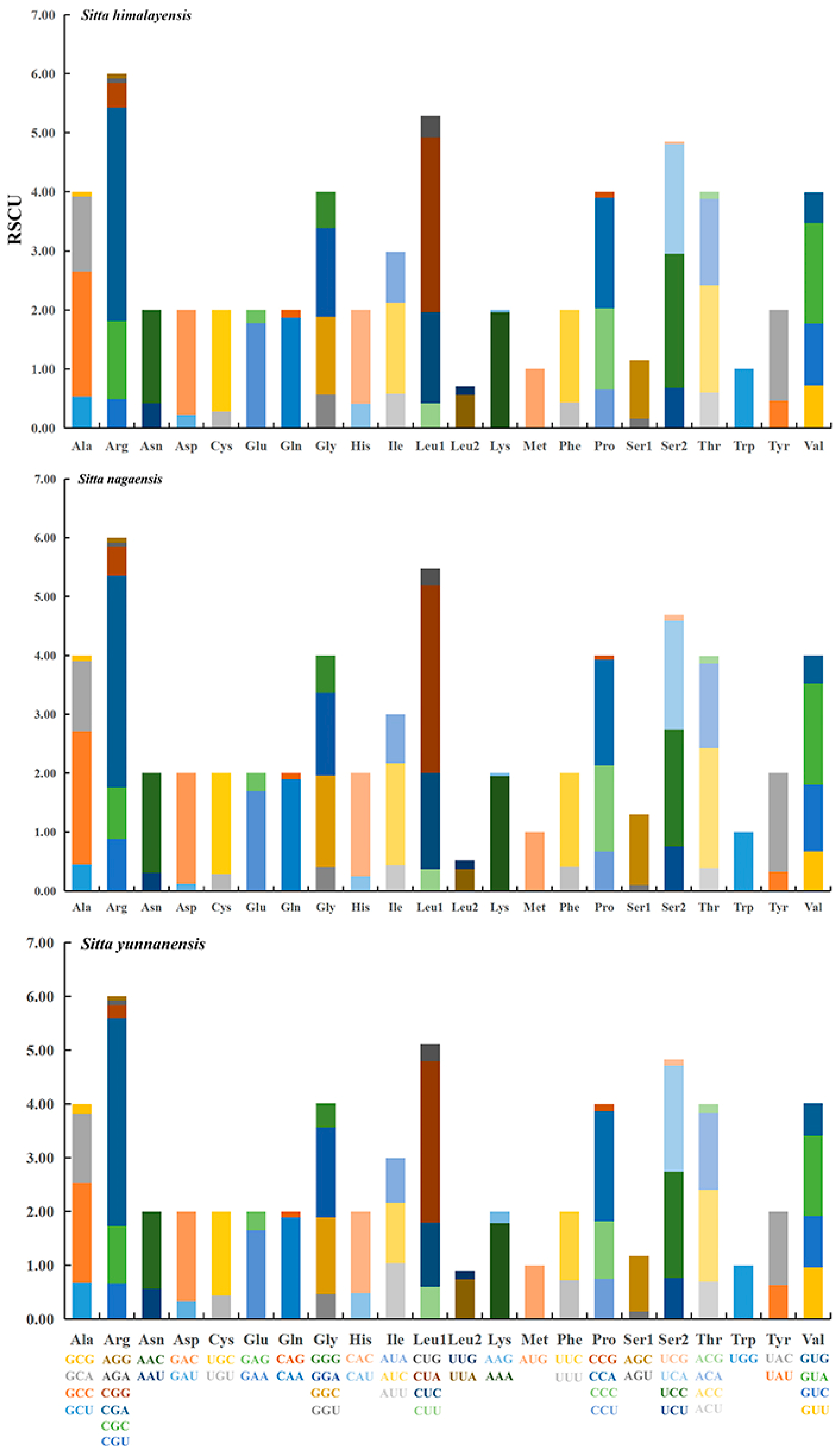
3.2. Protein-Coding Genes and Codon Usage
3.3. tRNA Genes and rRNA Genes
3.4. Control Region
3.5. Phylogenetic Analyses
4. Discussion
4.1. Mitogenome Characteristics
4.2. Phylogenetic Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.W.; Li, S.; Tian, C.Y.; Wang, Y.J.; Guo, Y.M. Molecular evolution and variability in mitochondrial DNA in ten species of Passeriforme. Acta Zool. Sin. 2002, 48, 625–632. [Google Scholar]
- Quinn, T.W. Molecular Evolution of the Mitochondrial Genome—Chapter 1. In Avian Molecular Evolution and Systematics; Academic Press: San Diego, CA, USA, 1997; pp. 3–28. [Google Scholar]
- Kan, X.Z.; Li, X.F.; Lei, Z.P.; Wang, M.; Chen, L.; Gao, H.; Yang, Z.Y. Complete mitochondrial genome of Cabot’s tragopan, Tragopan caboti (Galliformes: Phasianidae). Genet. Mol. Res. 2010, 9, 1204–1216. [Google Scholar] [CrossRef]
- Zhou, C.; Hao, Y.Q.; Ma, J.N.; Zhang, W.B.; Chen, Y.Z.; Chen, B.P.; Zhang, X.Y.; Yue, B.S. The first complete mitogenome of Picumnus innominatus (Aves, Piciformes, Picidae) and phylogenetic inference within the Picidae. Biochem. Syst. Ecol. 2017, 70, 274–282. [Google Scholar] [CrossRef]
- Sangster, G.; Luksenburg, J.A. Sharp increase of problematic mitogenomes of birds: Causes, consequences, and remedies. Genome Biol. Evol. 2021, 13, evab210. [Google Scholar] [CrossRef]
- HBW-BirdLife. Handbook of the Birds of the World and Birdlife, Version 2022-7. 2022. Available online: http://datazone.birdlife.org (accessed on 15 July 2022).
- Ericson, P.G.P.; Johansson, U.S. Phylogeny of Passerida (Aves: Passeriformes) based on nuclear and mitochondrial sequence data. Mol. Phylogenet. Evol. 2003, 29, 126–138. [Google Scholar] [CrossRef]
- Johansson, U.S.; Fjeldså, J.; Bowie, R.C. Phylogenetic relationships within Passerida (Aves: Passeriformes): A review and a new molecular phylogeny based on three nuclear intron markers. Mol. Phylogenet. Evol. 2008, 48, 858–876. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Alström, P.; Olsson, U.; Qu, Y.H.; Lei, F.M. Phylogenetic position of the Wallcreeper Tichodroma muraria. J. Ornithol. 2016, 157, 913–918. [Google Scholar] [CrossRef]
- Treplin, S.; Siegert, R.; Bleidorn, C.; Thompson, H.S.; Fotso, R.; Tiedemann, R. Molecular phylogeny of songbirds (Aves: Passeriformes) and the relative utility of common nuclear marker loci. Cladistics 2008, 24, 328–349. [Google Scholar] [CrossRef]
- Barker, F.K. Mitogenomic data resolve basal relationships among passeriform and passeridan birds. Mol. Phylogenet. Evol. 2014, 79, 313–324. [Google Scholar] [CrossRef]
- Chen, Z.T.; Du, Y.Z. First mitochondrial genome from Nemouridae (Plecoptera) reveals novel features of the elongated control region and phylogenetic implications. Int. J. Mol. Sci. 2017, 18, 996. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Zhou, L.Z.; Li, B.; Zhang, L.L. The complete mitochondrial genome of Aix galericulata and Tadorna ferruginea: Bearings on their phylogenetic position in the Anseriformes. PLoS One 2014, 9, e109701. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.C.; Cheng, Q.Q.; Ge, Y.S. The complete mitochondrial genome of Rhynchocypris oxycephalus (Teleostei: Cyprinidae) and its phylogenetic implications. Ecol. Evol. 2019, 9, 7819–7837. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.T.; Gong, L.; Zhang, Y.; Chen, J.; Liu, L.Q.; Jiang, L.H.; Lü, Z.M.; Liu, B.J.; Tong, G.X.; Wei, X.X. The complete mitochondrial genome of Calappa bilineata: The first representative from the family Calappidae and its phylogenetic position within Brachyura. Genomics 2020, 112, 2516–2523. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, W1, W54–W57. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Gary, B. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar]
- Oliveros, C.H.; Field, D.J.; Ksepka, D.T.; Barker, F.K.; Aleixo, A.; Andersen, M.J.; Alström, P.; Benz, B.W.; Braun, E.L.; Braun, M.J.; et al. Earth history and the passerine superradiation. Proc. Natl. Acad. Sci. USA 2019, 116, 7916–7925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stervander, M.; Fjeldså, J.; Christidis, L.; Ericson, P.G.P.; Ohlson, J.I.; Alström, P. An updated chronology of passerine birds. In The Largest Avian Radiation; Fjeldså, J., Christidis, L., Ericson, P.G.P., Eds.; Lynx Edicions: Barcelona, Spain, 2020; pp. 387–396. [Google Scholar]
- Zhang, Z.R.; Mi, S.H.; Guo, Q.X.; Zhang, Z.; Yan, P.F.; Liu, Z.S.; Teng, L.W. The complete mitochondrial genome of the Sitta villosa (Passeriformes: Sittidae) from China. Mitochondrial DNA Part B 2020, 5, 2328–2329. [Google Scholar] [CrossRef] [PubMed]
- Barker, F.K.; Oyler-McCance, S.; Tomback, D.F. Blood from a turnip: Tissue origin of low-coverage shotgun sequencing libraries affects recovery of mitogenome sequences. Mitochondrial DNA 2013, 26, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, C.; Léon, L.F.D.; Loaiza, J.R.; McMillan, W.O.; Miller, M.J. Extreme sequence divergence between mitochondrial genomes of two subspecies of White-breasted Wood-wren (Henicorhina leucosticte, Cabanis, 1847) from western and central Panamá. Mitochondrial DNA 2016, 27, 956–957. [Google Scholar] [CrossRef] [PubMed]
- Li, S.B.; Luo, A.; Li, G.P.; Li, W. Complete mitochondrial genome of the isabelline wheatear Oenanthe isabellina (Passeriformes, Muscicapidae). Mitochondrial DNA Part B 2016, 1, 355–356. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.F.; Yang, D.C.; Lu, C.H. Complete mitochondrial genome of oriental magpie-robin Copsychus saularis (Aves: Muscicapidae). Mitochondrial DNA Part B 2016, 1, 21–22. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Qin, J.H.; Luo, J.J.; Li, D.H.; Jiang, B.; Chang, C. Analysis of complete mitochondrial genome sequence of Kessleri thrush, Turdus kessleri (Passeriformes, Turdidae). Mitochondrial DNA Part B 2018, 3, 818–819. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.H.; Liu, B.; Lu, C.H. Complete mitochondrial genome of the Siberian thrush, Geokichla sibirica sibirica (Aves, Turdidae). Mitochondrial DNA Part B 2019, 4, 1150–1151. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. ClustalW and ClustalX version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.P.; Lin, Q.X.; Fang, W.Z.; Chen, X.L. The complete mitochondrial genomes of sixteen ardeid birds revealing the evolutionary process of the gene rearrangements. BMC Genom. 2014, 15, 573. [Google Scholar] [CrossRef] [Green Version]
- Santorum, J.M.; Darriba, D.; Taboada, G.L.; Posada, D. Jmodeltest.org: Selection of nucleotide substitution models on the cloud. Bioinformatics 2014, 30, 1310–1311. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; Mark, P.V.D.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F.; Nielsen, R.; Bollback, J.P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 2001, 294, 2310–2314. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J. Figtree Version 1.4.0. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 15 May 2021).
- Huang, Z.H.; Liao, X.J. Structure of the mitochondrial DNA control region and genetic variation of Chrysolophus pictus. Life Sci. Res. 2011, 15, 223–228. [Google Scholar]
- Xiao, B.; Ma, F.; Sun, Y.; Li, Q.W. Comparative analysis of complete mitochondrial DNA control region of four species of Strigiformes. Acta Genet. Sin. 2006, 33, 965–974. [Google Scholar] [CrossRef]
- Dejardins, P.; Morais, R. Sequence and gene organization of the chicken mitochondrial genome: A novel gene order in higher vertebrates. J. Mol. Biol. 1990, 212, 599–634. [Google Scholar] [CrossRef]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Boore, J.L. Complete mitochondrial genome sequence of the polychaete annelid Platynereis dumerilii. Mol. Biol. Evol. 2001, 18, 1413–1416. [Google Scholar] [CrossRef] [Green Version]
- Li, W.J.; Wang, Z.Q.; Che, Y.L. The complete mitogenome of the Wood-Feeding Cockroach Cryptocercus meridianus (Blattodea: Cryptocercidae) and its phylogenetic relationship among cockroach families. Int. J. Mol. Sci. 2017, 18, 2397. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.Y.; Xin, Z.Z.; Wang, Y.; Zhang, H.B.; Zhang, D.Z.; Wang, Z.F.; Zhou, C.L.; Tang, B.P.; Liu, Q.N. The complete mitochondrial genome of Clostera anachoreta (Lepidoptera: Notodontidae) and phylogenetic implications for Noctuoidea species. Genomics 2017, 109, 221–226. [Google Scholar] [CrossRef]
- Cracraft, J.; Barker, F.K.; Braun, M.; Harshman, J.; Dyke, G.J.; Feinstein, J.; Stanley, S.; Cibois, A.; Schikler, P.; Beresford, P.; et al. Phylogenetic relationships among modern birds (Neornithes): Towards an avian tree of life. In Assembling the Tree of Life; Cracraft, J., Donoghue, M., Eds.; Oxford University Press: Oxford, UK, 2004; pp. 468–489. [Google Scholar]
- Tobias, J.A.; Sheard, C.; Pigot, A.L.; Devenish, A.J.M.; Yang, J.Y.; Sayol, F.; Neate-Clegg, M.H.C.; Alioravainen, N.; Weeks, T.L.; Barber, R.A.; et al. A VONET: Morphological, ecological and geographical data for all birds. Ecol. Lett. 2022, 25, 581–597. [Google Scholar] [CrossRef]
- Päckert, M.; Bader-Blukott, M.; Künzelmann, B.; Sun, Y.H.; Hsu, Y.C.; Kehlmaier, C.; Albrecht, F.; Illera, J.C.; Martens, J. A revised phylogeny of nuthatches (Aves, Passeriformes, Sitta) reveals insight in intra- and interspecific diversification patterns in the Palearctic. Vertebr. Zool. 2020, 70, 241–262. [Google Scholar]
- Pasquet, E.; Barker, F.K.; Martens, J.; Tillier, A.; Cruaud, C.; Cibois, A. Evolution within the nuthatches (Sittidae: Aves, Passeriformes): Molecular phylogeny, biogeography, and ecological perspectives. J. Ornithol. 2014, 155, 755–765. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | GenBank No. | Mitogenome Size (bp) | Total A+T% | References |
|---|---|---|---|---|
| Sittidae | ||||
| Sitta nagaensis | MK343427 | 16,828 | 53.05 | This study |
| Sittaeuropaea | MN356255 | 16,827 | 53.19 | Unpublished |
| Sitta himalayensis | MK343426 | 16,822 | 53.91 | This study |
| Sittavillosa | MT444149 | 16,816 | 55.44 | [27] |
| Sittayunnanensis | MN052793 | 16,830 | 55.68 | This study |
| Sittacarolinensis | NC_024870 | 16,826 | 54.78 | [14] |
| Troglodytidae | ||||
| Campylorhynchus zonatus | NC_022840 | 16,780 | 50.57 | [28] |
| Campylorhynchus brunneicapillus | NC_029482 | 16,786 | 50.34 | Unpublished |
| Henicorhina leucosticta | NC_024673 | 16,727 | 52.88 | [29] |
| Muscicapidae | ||||
| Oenanthe isabellina | NC_040290 | 16,812 | 52.66 | [30] |
| Ficedula zanthopygia | JN018411 | 16,794 | 53.16 | Unpublished |
| Niltava davidi | NC_039538 | 16,770 | 54.16 | Unpublished |
| Cyornis hainanus/rubeculoides | HQ896033 | 16,802 | 53.01 | Unpublished |
| Copsychus saularis | NC_030603 | 16,827 | 52.74 | [31] |
| Turdidae | ||||
| Turdus kessleri | NC_041095 | 16,754 | 52.71 | [32] |
| Turdus eunomus | NC_028273 | 16,737 | 52.69 | Unpublished |
| Geokichla sibirica | MK377247 | 16,766 | 52.28 | [33] |
| Regulidae | ||||
| Regulus regulus | NC_029837 | 16,847 | 55.52 | Unpublished |
| Gene | Start Codon | Stop Codon | Anti-Codon | Strand | Intergenic Nucleotides (IGN) |
|---|---|---|---|---|---|
| trnF | GAA | H | −1 | ||
| rrnS | H | 0(a)/−1(b,c) | |||
| trnV | TAC | H | 21(a)/2(b,c) | ||
| rrnL | H | 2(a)/1(b)/7(c) | |||
| trnL2(UUR) | TAA | H | 11(a,c)/12(b) | ||
| nad1 | ATG | TAA/TA(A)/TAG | H | 7(a,c)/10(b) | |
| trnI | GAT | H | 6 | ||
| trnQ | TTG | L | 4(a)/3(b)/2(c) | ||
| trnM | CAT | H | 1(a,b)/0(c) | ||
| nad2 | ATG | TAA | H | 1 | |
| trnW | TCA | H | 1 | ||
| trnA | TGC | L | 10 | ||
| trnN | GTT | L | 2(a,c)/11(b) | ||
| trnC | GCA | L | −1 | ||
| trnY | GTA | L | 1 | ||
| cox1 | GTG | AGG | H | −9 | |
| trnS2(UCN) | TGA | L | 4 | ||
| trnD | GTC | H | 10(a)/11(b,c) | ||
| cox2 | ATG | TAA | H | 1 | |
| trnK | TTT | H | 1 | ||
| atp8 | ATG | TAA | H | −10 | |
| atp6 | ATG | TAA | H | 9(a,b)/11(c) | |
| cox3 | ATG | TA(A) | H | −1 | |
| trnG | TCC | H | 0 | ||
| nad3 | ATG | TAA | H | −1 | |
| trnR | TCG | H | 1 | ||
| nad4L | ATG | TAA | H | −7 | |
| nad4 | ATG | T(AA) | H | 0 | |
| trnH | GTG | H | 0 | ||
| trnS1(AGN) | GCT | H | −1 | ||
| trnL1(CUN) | TAG | H | 1(a)/0(b,c) | ||
| nad5 | ATG | AGA | H | 11 | |
| Cytb | ATG | TAA | H | 6(a)/3(b,c) | |
| trnT | TGT | H | 7(a)/8(b,c) | ||
| trnP | TGG | L | 14(a,b)/6(c) | ||
| nad6 | ATG | TAG | L | 0 | |
| trnE | TTC | L | 4(a)/5(b,c) | ||
| CR | H | 279(a)/282(b)/266(c) |
| Species | Whole Genome | PCGs | tRNAs | 16S rRNA | 12S rRNA | Control Region | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A+T (%) | AT Skew | GC Skew | A+T (%) | AT Skew | GC Skew | A+T (%) | AT Skew | GC Skew | A+T (%) | AT Skew | GC Skew | A+T (%) | AT Skew | GC Skew | A+T (%) | AT Skew | GC Skew | |
| S. himalayensis | 53.91 | 0.13 | −0.38 | 53.09 | 0.07 | −0.41 | 58.26 | 0.03 | 0.02 | 55.62 | 0.24 | −0.12 | 51.17 | 0.18 | −0.11 | 54.60 | −0.15 | −0.22 |
| S. nagaensis | 53.05 | 0.13 | −0.39 | 51.97 | 0.09 | −0.42 | 58.04 | 0.04 | 0.01 | 55.59 | 0.24 | −0.11 | 51.44 | 0.18 | −0.11 | 53.34 | −0.12 | −0.22 |
| S. yunnanensis | 55.68 | 0.10 | −0.35 | 55.17 | 0.04 | −0.38 | 58.31 | 0.04 | 0.01 | 56.32 | 0.23 | −0.10 | 52.25 | 0.18 | −0.11 | 55.49 | −0.15 | −0.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Q.; Guo, Q.; Cao, J.; Luo, X.; Duan, Y. Description of the Three Complete Mitochondrial Genomes of Sitta (S. himalayensis, S. nagaensis, and S. yunnanensis) and Phylogenetic Relationship (Aves: Sittidae). Genes 2023, 14, 589. https://doi.org/10.3390/genes14030589
Yuan Q, Guo Q, Cao J, Luo X, Duan Y. Description of the Three Complete Mitochondrial Genomes of Sitta (S. himalayensis, S. nagaensis, and S. yunnanensis) and Phylogenetic Relationship (Aves: Sittidae). Genes. 2023; 14(3):589. https://doi.org/10.3390/genes14030589
Chicago/Turabian StyleYuan, Qingmiao, Qiang Guo, Jing Cao, Xu Luo, and Yubao Duan. 2023. "Description of the Three Complete Mitochondrial Genomes of Sitta (S. himalayensis, S. nagaensis, and S. yunnanensis) and Phylogenetic Relationship (Aves: Sittidae)" Genes 14, no. 3: 589. https://doi.org/10.3390/genes14030589