
Comparative Chloroplast Genome Analyses of Six Hemlock Trees in East Asia: Insights into Their Genomic Characterization and Phylogenetic Relationship
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction and Sequencing
2.2. Genome Assembly, Annotation, and Sequence Analyses
2.3. Genome Comparison and Polymorphic Region Identification
2.4. Phylogenetic Analysis
3. Results
3.1. Genome Structure and Characteristics
3.2. Codon Usage Bias Analysis
3.3. SSR Analysis
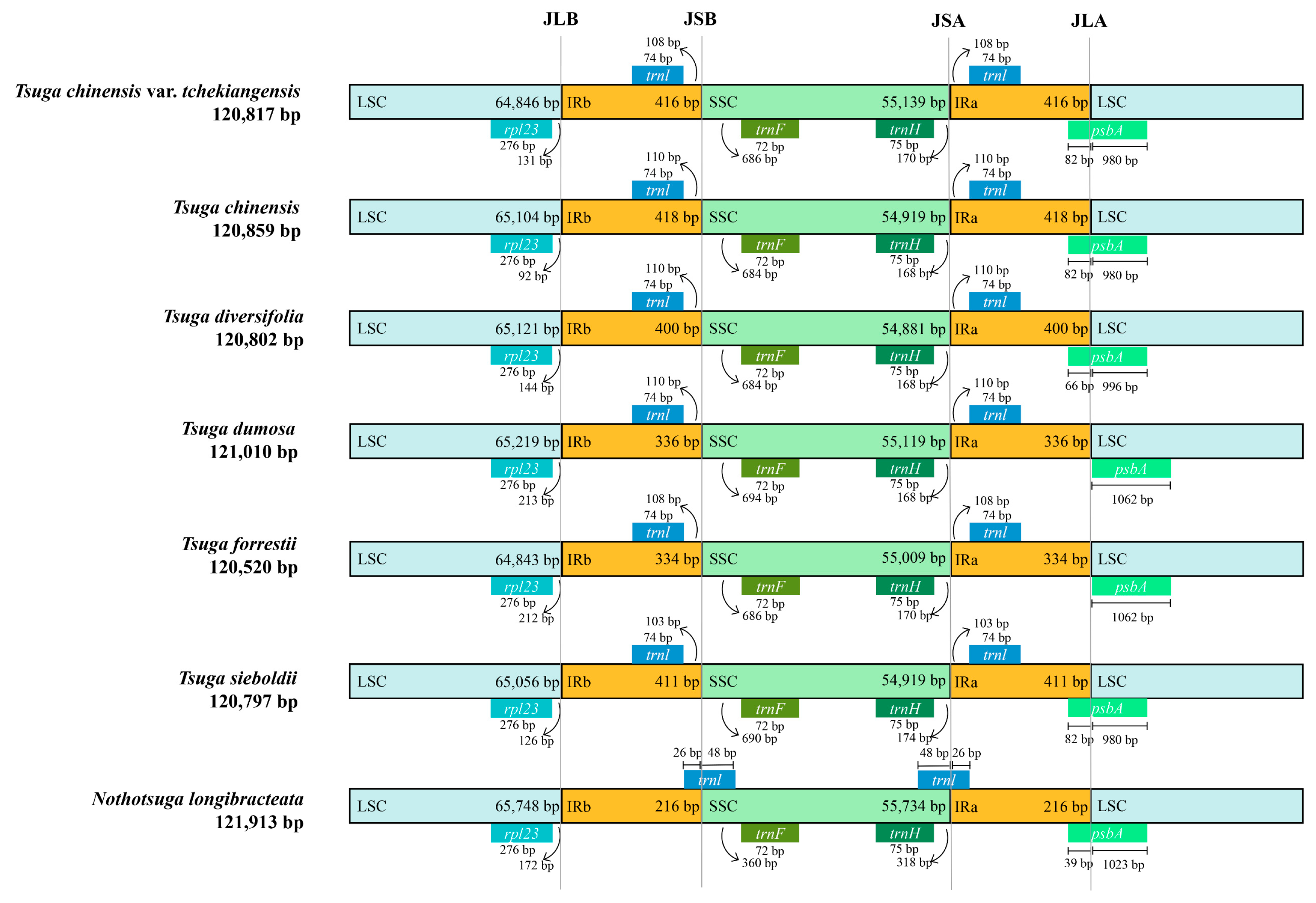
3.4. Comparison of IR Boundaries
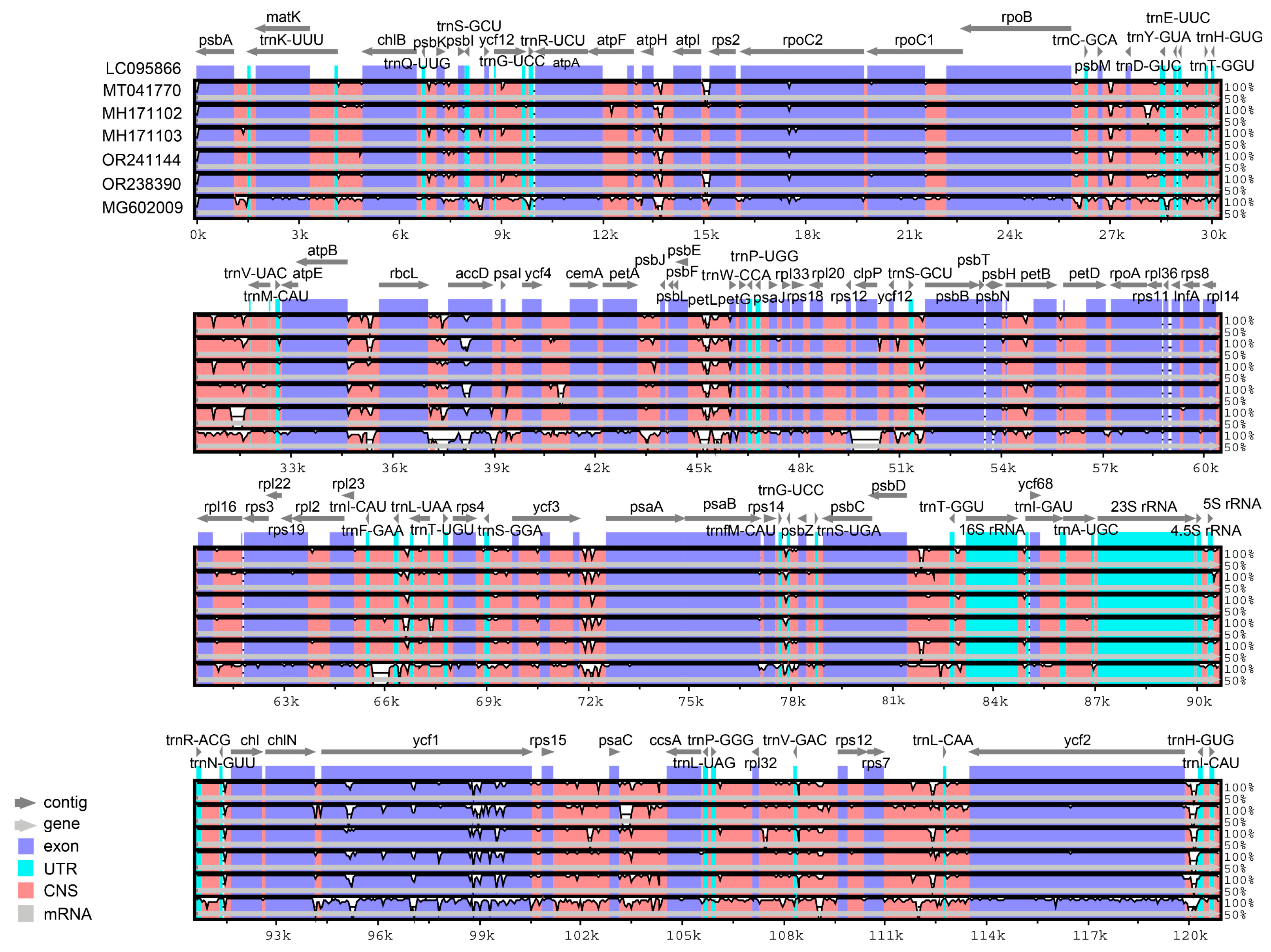
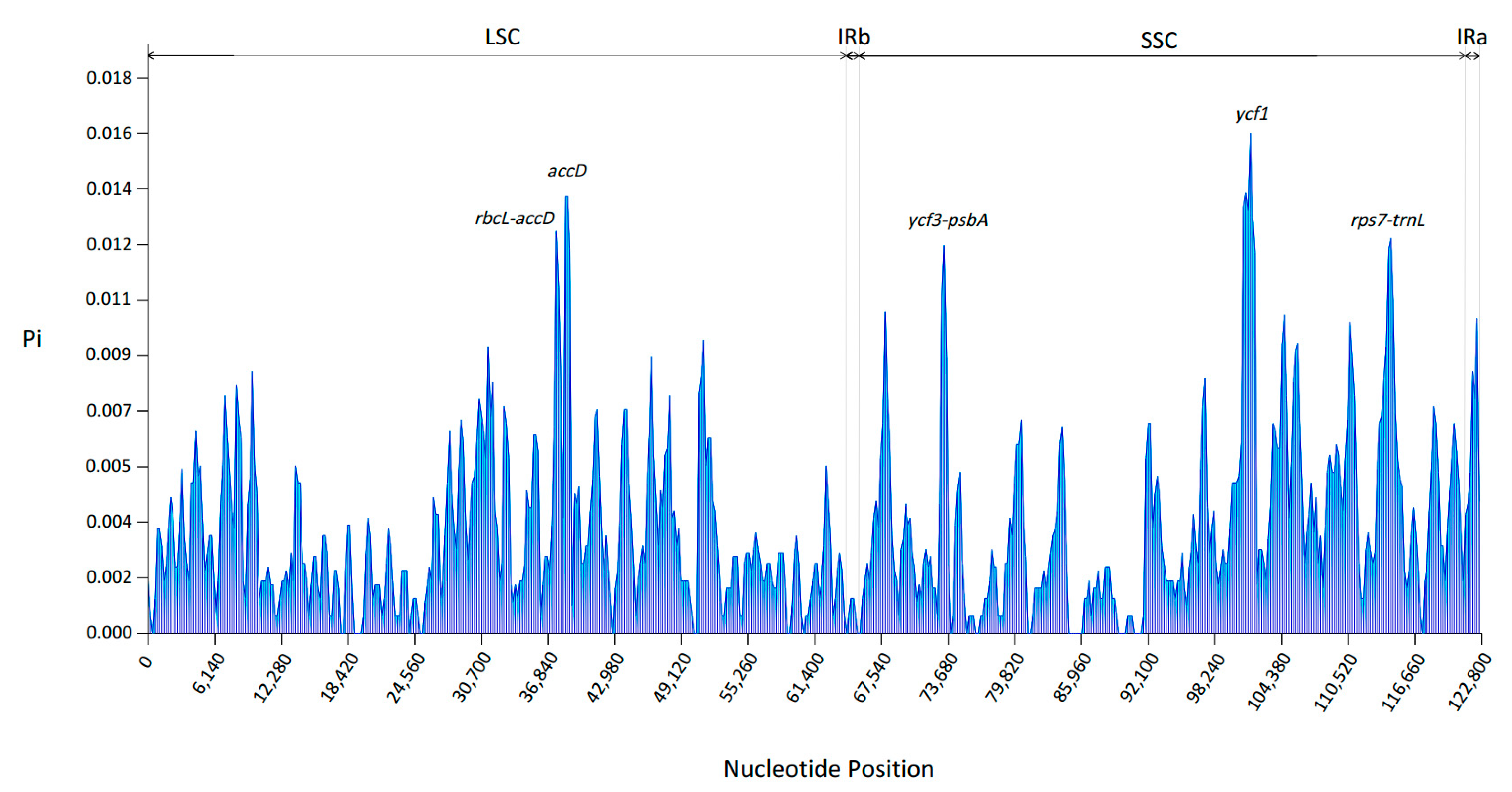
3.5. Sequence Divergence Analysis and Polymorphic Region Identification
3.6. Phylogenetic Relationships
4. Discussion
4.1. Characteristics of the Hemlock Chloroplast Genomes
4.2. The Phylogenetic Relationships of Tsuga and Related Genera
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Y.; Wang, Z.; Xu, X. Taxonomy and Distribution of Global Gymnosperms; Shanghai Scientific & Technical Publishers: Shanghai, China, 2017; pp. 638–650. [Google Scholar]
- Xiao, L.; Wang, H.; Guo, L.; Li, X.; Sun, N. Origin and spread of Tsuga (Endl.) Carrière based on fossil records. J. Earth Environ. 2023, 14, 9–26. [Google Scholar]
- Feng, Y.; Shen, T.; Shao, C.; Du, H.; Ran, J.; Wang, X. Phylotranscriptomics reveals the complex evolutionary and biogeographic history of the genus Tsuga with an East Asian-North American disjunct distribution. Mol. Phylogenet. Evol. 2021, 157, 107066. [Google Scholar] [CrossRef] [PubMed]
- Holman, G.; Del Tredici, P.; Havill, N.; Lee, N.S.; Cronn, R.; Cushman, K.; Mathews, S.; Raubeson, L.; Campbell, C.S. A new species and introgression in Eastern Asian hemlocks (Pinaceae: Tsuga). Syst. Bot. 2017, 42, 733–746. [Google Scholar] [CrossRef]
- Farjon, A. A Handbook of the World’s Conifers, 2nd ed.; Brill: Leiden-Boston, The Netherlands, 2017; pp. 1042–1055. [Google Scholar]
- Havill, N.P.; Campbell, C.S.; Vining, T.F.; Lepage, B.; Bayer, R.J.; Donoghue, M.J. Phylogeny and biogeography of Tsuga (Pinaceae) inferred from nuclear ribosomal ITS and chloroplast DNA sequence data. Syst. Bot. 2008, 33, 478–489. [Google Scholar] [CrossRef]
- Cun, Y.Z.; Wang, X.Q. Phylogeography and evolution of three closely related species of Tsuga (hemlock) from subtropical eastern Asia: Further insights into speciation of conifers. J. Biogeogr. 2015, 42, 315–327. [Google Scholar] [CrossRef]
- Cheng, W.C.; Fu, L.K. Tsuga. In Flora Reipublicae Popularis Sinicae; Science Press: Beijing, China, 1978; Volume 7, p. 119. [Google Scholar]
- Fu, L.; Li, N.; Mill, R.R. Pinaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 1999; Volume 4, pp. 11–52. [Google Scholar]
- Liu, L.; Li, P.; Zhang, H.; Worth, J. Whole chloroplast genome sequences of the Japanese hemlocks, Tsuga diversifolia and T. sieboldii, and development of chloroplast microsatellite markers applicable to East Asian Tsuga. J. For. Res. 2018, 23, 318–323. [Google Scholar] [CrossRef]
- Mogensen, H.L. The Hows and Whys of cytoplasmic inheritance in seed plants. Am. J. Bot. 1996, 83, 383–404. [Google Scholar] [CrossRef]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Depamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar]
- Wang, X.; Tank, D.C.; Sang, T. Phylogeny and divergence times in Pinaceae: Evidence from three genomes. Mol. Biol. Evol. 2000, 17, 773–781. [Google Scholar] [CrossRef]
- Kormutak, A.; Galgoci, M.; Manka, P.; Koubova, M.; Jopcik, M.; Sukenikova, D.; Bolecek, P.; Gőmőry, D. Field-based artificial crossings indicate partial compatibility of reciprocal crosses between Pinus sylvestris and Pinus mugo and unexpected chloroplast DNA inheritance. Tree Genet. Genomes 2017, 13, 68. [Google Scholar] [CrossRef]
- Zhao, Y. Progress in the application of chloroplast genome in botanical research. Biol. Teach. 2022, 47, 83–85. [Google Scholar]
- Chen, L.; Li, L.; Yang, G.; Qian, H.; Li, M. Characterization of the complete chloroplast genome sequence of Tsuga longibracteata W. C. Cheng (Pinaceae). Conserv. Genet. Resour. 2019, 11, 117–120. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids. Res. 2016, w955. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids. Res. 2013, 41, w575–w581. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids. Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids. Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2. Wires Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S.; Program, N.C.S. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids. Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Delbarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Tanner-Smith, E.E.; Tipton, E. Robust variance estimation with dependent effect sizes: Practical considerations including a software tutorial in Stata and SPSS. Res. Synth. Methods 2014, 5, 13–30. [Google Scholar] [CrossRef]
- Almutairi, M.M. Analysis of chromosomes and nucleotides in rice to predict gene expression through codon usage pattern. Saudi J. Biol. Sci. 2021, 28, 4569–4574. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Cai, G.; Cai, R.; Hu, Z.; Wang, H. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids. Res. 2023, 51, w587–w592. [Google Scholar] [CrossRef]
- Sudianto, E.; Wu, C.; Lin, C.; Chaw, S. Revisiting the plastid phylogenomics of Pinaceae with two complete plastomes of Pseudolarix and Tsuga. Genome Biol. Evol. 2016, 8, 1804–1811. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ferguson, D.K.; Liu, B.; Mao, K.; Gao, L.; Zhang, S.; Wan, T.; Rushforth, K.; Zhang, Z. Recent advances on phylogenomics of gymnosperms and a new classification. Plant Divers. 2022, 44, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Ye, Y.; Bai, T.; Xu, M.; Xu, L. Complete chloroplast genome of Pinus massoniana (Pinaceae): Gene rearrangements, loss of ndh genes, and short inverted repeats contraction, expansion. Molecules 2017, 22, 1528. [Google Scholar] [CrossRef] [PubMed]
- Ping, J.; Hao, J.; Li, J.; Yang, Y.; Su, Y.; Wang, T. Loss of the IR region in conifer plastomes: Changes in the selection pressure and substitution rate of protein-coding genes. Ecol. Evol. 2022, 12, e8499. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, M.; Zhang, C.; Xiang, S.; Zheng, H.; He, Y. Comparative study on chloroplast genome and phylogenetic analysis of Abies. Biol. Chem. Eng. 2021, 7, 56–60. [Google Scholar]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.; Gao, L. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, X.; Tang, X.; Zhang, C. Endangered species of Pseudotsuga brevifolia chloroplast genome characteristics and Pinaceae species phylogenetic analysis. Mol. Plant Breed. 2021, 21, 3917–3918. [Google Scholar]
- Bulmer, M. The selection-mutation-drift theory of synonymous codon usage. Genetics 1991, 129, 897–907. [Google Scholar] [CrossRef]
- Wu, P.; Xiao, W.; Luo, Y.; Xiong, Z.; Chen, X.; He, J.; Sha, A.; Gui, M.; Li, Q. Comprehensive analysis of codon bias in 13 Ganoderma mitochondrial genomes. Front. Microbiol. 2023, 14, 1170790. [Google Scholar] [CrossRef]
- Kawabe, A.; Miyashita, N.T. Patterns of codon usage bias in three dicot and four monocot plant species. Genes Genet. Syst. 2003, 78, 343–352. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Dong, H.; Jiang, C.; Cao, F.; Tao, S.; Xu, L. Analysis of codon usage patterns in Ginkgo biloba reveals codon usage tendency from A/U-ending to G/C-ending. Sci. Rep. 2016, 6, 35927. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Luo, Y.; Liu, X.; Lu, X.; Yin, G.; Deng, Z. Synonymous codon usage bias in the chloroplast genome of Metasequoia glyptostroboides. J. Hubei Minzu Univ. (Nat. Sci. Ed.) 2022, 40, 121–128. [Google Scholar]
- Zheng, W.; Chen, J.; Hao, Z.; Shi, J. Analysis on codon usage bias of chloroplast genes from five conifers including Cunninghamia lanceolata. Mol. Plant Breed. 2016, 14, 1091–1097. [Google Scholar]
- Kuang, D.; Wu, H.; Wang, Y.; Gao, L.; Zhang, S.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef]
- Li, L.; Wu, Q.; Fang, L.; Wu, K.; Li, M.; Zeng, S. Comparative chloroplast genomics and phylogenetic analysis of Thuniopsis and closely related genera within Coelogyninae (Orchidaceae). Front. Genet. 2022, 13, 850201. [Google Scholar] [CrossRef]
- Moghaddam, M.; Ohta, A.; Shimizu, M.; Terauchi, R.; Kazempour-Osaloo, S. The complete chloroplast genome of Onobrychis gaubae (Fabaceae-Papilionoideae): Comparative analysis with related IR-lacking clade species. BMC Plant Biol. 2022, 22, 75. [Google Scholar] [CrossRef]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef]
- Wu, C.; Lin, C.; Hsu, C.; Wang, R.; Chaw, S. Comparative chloroplast genomes of Pinaceae: Insights into the mechanism of diversified genomic organizations. Genome Biol. Evol. 2011, 3, 309–319. [Google Scholar] [CrossRef]
- Ran, J.; Shen, T.; Wu, H.; Gong, X.; Wang, X. Phylogeny and evolutionary history of Pinaceae updated by transcriptomic analysis. Mol. Phylogenet. Evol. 2018, 129, 106–116. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | T. chinensis var. tchekiangensis | T. chinensis | T. diversifolia | T. dumosa | T. forrestii | T. sieboldii |
|---|---|---|---|---|---|---|
| GenBank No. | MT041770 | LC095866 [35] | MH171102 [10] | OR241144 * | OR238390 * | MH171103 [10] |
| Size (bp’s) | 120,817 | 120,859 | 120,802 | 121,010 | 120,520 | 120,797 |
| LSC (bp’s) | 64,846 | 65,104 | 65,121 | 65,219 | 64,843 | 65,056 |
| SSC (bp’s) | 55,139 | 54,919 | 54,881 | 55,119 | 55,009 | 54,919 |
| IR (bp’s) | 416 | 418 | 400 | 336 | 334 | 411 |
| Coding (bp’s) | 68,756 | 68,837 | 62,453 | 61,764 | 61,653 | 68,794 |
| Noncoding (bp’s) | 52,061 | 52,002 | 58,349 | 59,246 | 58,867 | 52,003 |
| Number of genes | 111 | 112 | 111 | 108 | 108 | 111 |
| Protein-coding genes | 72 | 73 | 72 | 72 | 72 | 72 |
| tRNA genes | 35 | 35 | 35 | 32 | 32 | 35 |
| rRNA genes | 4 | 4 | 4 | 4 | 4 | 4 |
| Total GC (%) | 38.1 | 38.1 | 38.1 | 38.2 | 38.1 | 38.1 |
| LSC (%) | 37.4 | 37.4 | 37.4 | 37.4 | 37.5 | 37.4 |
| SSC (%) | 38.9 | 38.9 | 39 | 39 | 39.1 | 38.9 |
| IR (%) | 37.2 | 37.3 | 37.3 | 37.5 | 35.9 | 36 |
| Class Boundary | T. chinensis var. tchekiangensis | T. chinensis | T. diversifolia | T. dumosa | T. forrestii | T. sieboldii | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | F/(%) | No. | F/(%) | No. | F/(%) | No. | F/(%) | No. | F/(%) | No. | F/(%) | |
| −0.15~−0.05 | 1 | 2.27 | 1 | 2.27 | 1 | 2.32 | 1 | 2.27 | 1 | 2.27 | 1 | 2.27 |
| −0.05~0.05 | 12 | 27.28 | 12 | 27.28 | 12 | 27.91 | 12 | 27.28 | 12 | 27.28 | 12 | 27.28 |
| 0.05~0.15 | 21 | 47.73 | 21 | 47.73 | 20 | 46.52 | 21 | 47.73 | 21 | 47.73 | 21 | 47.73 |
| 0.15~0.25 | 9 | 20.45 | 9 | 20.45 | 9 | 20.93 | 9 | 20.45 | 9 | 20.45 | 9 | 20.45 |
| 0.25~0.35 | 1 | 2.27 | 1 | 2.27 | 1 | 2.32 | 1 | 2.27 | 1 | 2.27 | 1 | 2.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Liu, X.; Wang, Z.; Wu, X.; Hong, K.; Xie, C. Comparative Chloroplast Genome Analyses of Six Hemlock Trees in East Asia: Insights into Their Genomic Characterization and Phylogenetic Relationship. Forests 2023, 14, 2136. https://doi.org/10.3390/f14112136
Chen L, Liu X, Wang Z, Wu X, Hong K, Xie C. Comparative Chloroplast Genome Analyses of Six Hemlock Trees in East Asia: Insights into Their Genomic Characterization and Phylogenetic Relationship. Forests. 2023; 14(11):2136. https://doi.org/10.3390/f14112136
Chicago/Turabian StyleChen, Lin, Xin Liu, Zhibei Wang, Xi Wu, Kaiyue Hong, and Chunping Xie. 2023. "Comparative Chloroplast Genome Analyses of Six Hemlock Trees in East Asia: Insights into Their Genomic Characterization and Phylogenetic Relationship" Forests 14, no. 11: 2136. https://doi.org/10.3390/f14112136