 Qingchao Zeng
Qingchao Zeng Xiaowu Man
Xiaowu Man Annie Lebreton
Annie Lebreton Yucheng Dai1,2*
Yucheng Dai1,2* Francis M. Martin
Francis M. Martin- 1Beijing Advanced Innovation Center for Tree Breeding by Molecular Design, Beijing Forestry University, Beijing, China
- 2School of Ecology and Nature Conservation, Institute of Microbiology, Beijing Forestry University, Beijing, China
- 3College of Forestry, Beijing Forestry University, Beijing, China
- 4Université de Lorraine, INRAE, UMR Interactions Arbres/Microorganismes, Centre INRAE, Grand Est-Nancy, Champenoux, France
Ectomycorrhizal (ECM) symbioses play an important role in tree biology and forest ecology. However, little is known on the composition of bacterial and fungal communities associated to ECM roots. In the present study, we surveyed the bacterial and fungal microbiome of ECM roots from stone oaks (Lithocarpus spp.) and Yunnan pines (Pinus yunnanensis) in the subtropical forests of the Ailao Mountains (Yunnan, China). The bacterial community was dominated by species pertaining to Rhizobiales and Acidobacteriales, whereas the fungal community was mainly composed of species belonging to the Russulales and Thelephorales. While the bacterial microbiome hosted by ECM roots from stone oaks and Yunnan pines was very similar, the mycobiome of these host trees was strikingly distinct. The microbial networks for bacterial and fungal communities showed a higher complexity in Lithocarpus ECM roots compared to Pinus ECM roots, but their modularity was higher in Pinus ECM roots. Seasonality also significantly influenced the fungal diversity and their co-occurrence network complexity. Our findings thus suggest that the community structure of fungi establishing and colonizing ECM roots can be influenced by the local soil/host tree environment and seasonality. These results expand our knowledge of the ECM root microbiome and its diversity in subtropical forest ecosystems.
Introduction
Boreal, temperate and tropical forests are major terrestrial biomes and host a large part of the plant, animal and microbial diversity (Baldrian, 2017). Importantly, forests play a crucial role in global climate regulation (Pan et al., 2011; Pohjanmies et al., 2017; Mishra et al., 2020). Forest trees are tightly associated to soil microorganisms, including widespread beneficial mycorrhizal associations (van der Heijden et al., 2015). Ectomycorrhizal (ECM) fungi colonize up to 95% of tree root tips in temperate, boreal and subtropical forest ecosystems—biomes that make up much of the global terrestrial carbon sink. These root-colonizing fungi receive photosynthetically-fixed carbon from their host plants in exchange for plant growth-limiting soil nutrients, such as nitrogen and phosphorus, and they are key players in soil carbon sequestration. Apart from improving tree nutrition, symbiotic fungi also enhance plant resistance against abiotic and biotic stresses (van der Heijden et al., 2015; Nehls and Plassard, 2018; Dreischhoff et al., 2020). Together with other commensal and endophytic bacteria and fungi, ECM symbionts constitute the plant microbiome which significantly expands the genomic potential of the host as it serves functions that support host development, growth and nutrition (Berendsen et al., 2012; Turner et al., 2013; Berg et al., 2014; Beckers et al., 2017). Mycorrhizal roots represent an important niche for soil bacteria and fungi (Garbaye, 1994; Perotto and Bonfante, 1997; Bending et al., 2002; Frey Klett et al., 2007; Marupakula et al., 2016). However, the factors influencing the composition and structure of the microbial communities inhabiting ECM roots and their interactions are still poorly known. Given the importance of ECM fungi in tree biology and forest ecology, there is thus a need for a better understanding of the mechanisms driving the assembly of bacterial and fungal communities in ECM roots.
Izumi et al. (2007) identified that the most abundant cultivable bacteria found in Pinus nigra roots colonized by the fungal ECM fungi Suillus variegatus and Tomentellopsis submollis pertained to the Pseudomonas, Burkholderia and Bacillus genera. Further studies have shown that the zone adjacent to hyphal tips of arbuscular mycorrhizal fungi (AMF), the so-called hyphosphere, hosted a wide range of bacteria belonging to the Firmicutes, Actinobacteria, Bacteroidetes, Proteobacteria, Chloroflexi, Planctomycetes and Verrucomicrobia (Marupakula et al., 2016; Emmett et al., 2021). Bacterial populations were much higher in the rhizosphere compared to the hyphosphere and a striking shift in the bacterial composition between the hyphosphere and the rhizosphere has been reported (Andrade et al., 1997; Frey Klett et al., 2007). Bacillus and Arthrobacter were frequently found in the hyphosphere, while Pseudomonas dominated in the rhizosphere soil confirming the effect of mycorrhizal fungi on bacterial composition (Frey Klett et al., 2007; Gahan and Schmalenberger, 2015; Zhang et al., 2021). In Betula pubescens, it has also been shown that different ECM associations hosted distinctive bacterial and fungal microbiomes (Izumi and Finlay, 2011). Carbon compounds, such as trehalose, released by the fungal mycelium likely promote bacterial growth and the mycelial networks may facilitate bacterial co-migration in soil (Frey Klett et al., 2007; Nazir et al., 2010; Warmink et al., 2011). On the other hand, the direct access of ECM fungi to glucose released by root cells entails a competitive advantage over other soilborne microbes. Thus, the presence of ECM fungi can, in some circumstances, lead to suppressed growth and respiration by other soil microorganisms, such as saprotrophic fungi, thereby further increasing belowground carbon sequestration. In turn, bacteria have beneficial effects on symbiotic fungi; Burkholderia BS001 is able to protect its fungal host from the detrimental effect of antifungal agents (Nazir et al., 2014), whereas some mycorrhiza-associated bacteria produce compounds that are antagonistic to plant pathogens (Riedlinger et al., 2006; Frey Klett et al., 2007). Pseudomonas and Bacillus of the AMF hyphosphere mobilize soil inorganic phosphorus (Wang et al., 2016). Mycorrhizal helper bacteria play a role in diverse key functions, including germination of fungal propagules, promotion of mycelial growth, reduction of soil-mediated stresses and affect host recognition (Garbaye, 1994; Perotto and Bonfante, 1997; Bending et al., 2002; Frey Klett et al., 2007; Aspray et al., 2013; Marupakula et al., 2016; Zhang et al., 2021).
Biotic and abiotic factors strongly influence the composition of bacterial and fungal communities. Plant developmental stages shape the assembly patterns of the plant microbiome (Xiong et al., 2021a), including the community of ECM fungi. The composition of the microbiome inhabiting ECM roots is modulated by the host tree genotype and physiology, but also by rhizospheric microorganisms, soil properties and seasonality (Frey Klett et al., 2007; Izumi and Finlay, 2011; Bonito et al., 2014; Deveau, 2016). Analysis of the core bacterial microbiome of ECM roots suggested that it is not influenced by the species of ECM fungi, while roots play an important role in shaping its composition (Marupakula et al., 2016). However, the contribution and importance of tree species on the ECM microbiome composition remain poorly understood. A deeper knowledge of the mechanisms that govern the assemblage of microbial communities in ECM roots is needed to better understand and predict the greater ecosystem impacts of ECM associations. This includes determining how the host affects the microbiome composition, as well as how ECM fungi impact their associated microbes.
In a companion study, we surveyed by metabarcoding sequencing, the diversity and composition of soil bacteria and fungi in an old-growth forest, dominated by stone oaks (Lithocarpus spp.) and in a secondary Yunnan pine woodland in the subtropical Ailao Mountains in the Yunnan province, China (Zeng et al., 2022). Limited information exists on the alterations of soil microbiological characteristics in subtropical montane forests, while little is known of how the microbiome distribution pattern, life history and functional traits vary during forest replacement, e.g., old-growth Lithocarpus forest versus native pine woodlands. We therefore assessed the effect of forest replacement and other environmental factors, including soil horizons, soil physicochemical characteristics and seasonality (monsoon vs. dry seasons). We showed that tree composition and variation in soil properties were major drivers for both soil bacterial and fungal communities, with a significant influence from seasonality. ECM symbionts dominated the functional fungal guilds. Owing to the importance of the ECM fungal community, we aimed here to characterize the fungal and bacterial microbiomes of ECM roots from stone oaks and Yunnan pines. We have used culture-independent, high-throughput ribosomal DNA amplicon sequencing to investigate variation patterns in bacterial and fungal communities associated to ECM roots. We hypothesized that (1) tree species would be the primary factor shaping the ECM root microbiome due to host selection effects and (2) the microbial composition would differ with environmental factors, such as seasonality.
Materials and methods
Experimental design and sample collection
The sampling site was located in the Ailaoshan National Forest Ecosystem Research Station in Jingdong country, Yunnan Province (24°31′ N, 101°01′ E). The Ailaoshan lie between the southern and northern subtropical forest formations in a transition area. The most extensive forest ecosystem is a contiguous primary, old-growth broadleaf evergreen stone oaks (Lithocarpus) association which covers 75–80% of the region (Young et al., 1992). Two types of forests, dominated by Lithocarpus sp. and Pinus yunnanensis, were selected in this study. The stone oaks (Lithocarpus) old-growth forest, lies in a protected area of 5,100 ha of evergreen forest with a stand age of more than 300 years (Song et al., 2017). The upper canopy of the forest is 18 to 25 m high. Diameter at breast height (DBH) of the selected stone oaks ranged from 75 to 180 cm. The soils are loamy Alfisols (Chan et al., 2006; Wen et al., 2017). The nearby Pinus woodland replaced evergreen old-growth broadleaf stands about 55–65 years ago (Young and Wang, 1989). The pine woodland was located in an open, park-like area with a minimum of human interference. The DBH of the selected pine trees ranged from 65 to 95 cm. Although these two forest associations are located a dozens of km apart, they had few plant species in common. In this area, the annual mean temperature is 11.6°C and the mean annual precipitation is 1799 mm, 86% of which occurs during the wet season from May to October (Song et al., 2017). Sample collection in these two forests was performed in April and August 2020, corresponding to the end of the dry season and wet seasons, respectively.
Both stone oaks and Yunnan pines are known to form ECM associations. We collected ECM root samples in six plots, three located beneath stone oaks and three located beneath Yunnan pines. Individual plots were separated by ~100 m and the area of each plot was ~900 m2. In each plot, four individual adult trees, separated by 5 m to 15 m, were selected for ECM root collection. For each tree, two samples were collected at 1 m (north and south) from the base of the trunk. We sampled the ECM root tips and ECM root cluster found in a series of soil samples (20 × 20 cm, depth of 0–30 cm). In total, 96 samples were collected for microbial community DNA analysis (2 sites × 3 plots × 4 trees × 2 directions × 2 seasons). ECM root tips and clusters were separated from the main root system using forceps and a cutter, and any remaining soil particles attached to ECM roots or ECM root clusters (Supplementary Figure S1) were carefully removed using forceps and distilled water. ECM roots were distinguished based on external morphological characteristics (i.e., presence of a fungal mantle, forked pine roots, clustered roots). Between 10 to 100 ECM roots were harvested per replicate and snap frozen in dry ice until brought back to the Kunming Institute of Botany (KIB) where root samples were stored at −80°C prior to DNA extraction.
DNA extraction, gene amplification and sequencing
Total DNA was extracted from ECM roots using a modified CTAB protocol (Maropola et al., 2015). In brief, 850 μl CTAB buffer (1 M Tris–HCl, 4 M NaCl, 0.5 M EDTA, 2% CTAB, 0.2% β-mercaptoethanol) was added to ECM roots and incubated at 65°C for 1 h, followed by the addition of 850 μl 24:1 (v/v) chloroform/isoamyl alcohol solution. The tubes were vortexed for 10 min and centrifuged (12,000 rpm, 10 min). The supernatant was collected in a new tube and 850 μl chloroform/isoamyl solution was added to the samples. Next, the tube was vortexed for 10 min and centrifuged (12,000 rpm, 10 min). Equal volumes of ice-cold isopropanol were added to the supernatants collected in a fresh tube and mixed by inverting, followed by incubation at-20°C for 24 h and centrifugation (12,000 rpm, 30 min). The supernatants were discarded and DNA pellets were washed twice with 800 μl 75% ethanol and eluted following centrifugation (12,000 rpm, 3 min). The DNA pellets were air-dried and then resuspended in 80 μl ddH2O before storing at-20°C. The concentration and quality of DNA were determined using the Nanodrop ONE spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States).
Bacterial and fungal communities were analyzed by high-throughput DNA metabarcoding sequencing. Bacterial communities were profiled by targeting a region of the 16S rRNA using the primer pairs 515F/806R. The composition of the fungal community was assessed by targeting the internal transcribed spacers (ITS). The ITS1 primers were used to amplify the samples collected during the dry and wet seasons, whereas the 16S and ITS2 primers were only used to amplify the samples collected during the wet season. We sequenced both the ITS1 and ITS2 regions using the primer pairs ITS1F/ITS2 and ITS86/ITS4, respectively (Caporaso et al., 2011; De Beeck et al., 2014). All PCR reactions were performed in 30 μl volume with 15 μl of Phusion® high-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM each of forward and reverse primers and 10 ng template DNA. PCR amplifications were performed using the following program: 1 min initial denaturation at 98°C, 30 cycles of 10 s at 98°C, 30 s at 50°C, and 30 s at 72°C, with a final 5 min elongation at 72°C. Libraries were generated using the Illumina TruSeq DNA PCR-Free Library Preparation Kit (Illumina, United States) following the manufacturer’s recommendations and index codes were added. The library quality was assessed using the Qubit 2.0 Fluorometer (Thermo Scientific) and on the Agilent Bioanalyzer 2,100 system. All samples were pooled in equimolar concentrations and subsequently sequenced on the Illumina NovaSeq platform with a paired-end protocol by Novogene Biotech Co., Ltd. (Beijing, China).
Bioinformatic processing of the sequences
Amplicon data were analyzed using a combination of VSEARCH v2.13.3 and QIIME 1.9.1. softwares (Caporaso et al., 2010; Zgadzaj et al., 2016). Raw sequences were split based on their unique barcodes and the primer sequences, and low-quality sequences were trimmed off using an in-house script. Next, the paired-end sequences were merged using the USEARCH software v11.0.667 and the resultant sequences were quality-filtered. Singletons were removed using VSEARCH. The sequence reads were then clustered into operational taxonomic units (OTUs) at 97% similarity level using the UPARSE pipeline. Chimeric sequences that were identified using the reference-based methods were removed from the data. Representative sequences were classified using the BLAST algorithm with SILVA v.13.2 and UNITE v8.0 reference databases (Quast et al., 2013; Nilsson et al., 2019). Mitochondrial and chloroplast DNA sequences, as well as the OTUs with a total relative abundance of <0.00001 in all samples were discarded. The raw sequencing data have been submitted to the Sequence Read Archive under the accession number PRJNA820166 (16S and ITS2) and PRJNA782391 (ITS1).
Statistical analysis
To assess alpha-diversity indices, a rarefaction step of sequence reads was performed to obtain the same amount of reads among samples, i.e., 29,715, 34,997 and 6,744 reads, corresponding to the lowest number of sequenced reads per sample, for 16S, ITS1 and ITS2 amplicons, respectively. The cumulative sum scaling (CSS) was used as a normalization method for beta-diversity analysis. A principal coordinate analysis (PCoA) was performed using the cmdscale function in the vegan packages. Bacterial and fungal alpha-diversity (observed OTUs and Shannon index) was calculated using QIIME. The observed significant differences were evaluated by one-way ANOVA (analysis of variance). Bacterial and fungal beta-diversity was estimated according to the Bray-Curtis distance between samples. Permutational multivariate ANOVA (PERMANOVA) statistical tests were performed using the R packages, vegan, with the adonis function having 999 or 1,000 permutations (Xiong et al., 2021b).
Venn diagrams were drawn using the OECloud tools (https://cloud.oebiotech.cn) to analyze the overlapping and unique OTUs between Lithocarpus and Pinus samples. Differential abundance between Lithocarpus and Pinus samples was assessed at phylum and class levels using the STAMP software and Welch’s tests followed by Benjamini-Hochberg FDR corrections (Parks et al., 2014). This analysis was also performed at OTUs level using the Edge’s generalized linear model (GLM) in the “edgeR” packages, with trimmed mean of M-values (TMM) normalization method and a threshold of significance set at p < 0.05 (Robinson et al., 2010). To identify the biomarkers of different tree species and sampling season, a linear discriminant analysis effect size (LEfSe) was employed (Wilcoxon value of p <0.05, logarithmic LDA (linear discriminant analysis) score > 2; http://huttenhower.sph.harvard.edu/galaxy; Segata et al., 2011). To elucidate the microbial interactions taking place in the two forest associations and at different sampling seasons, microbial association networks were created using the OTU tables. To reduce network complexity, OTUs present in all samples for bacterial and 40% or 60% samples for fungal communities were selected to construct the co-occurrence networks (Xiong et al., 2021b). Spearman’s correlation coefficient between two OTUs were considered statistically robust at ρ > 0.6 with a corresponding value of p of <0.01 (Barberan et al., 2014). The pairwise comparisons based on abundances were performed using the rcorr function in the “Hmisc” package and the value of p was adjusted using the Benjamini-Hochberg method (Zhu et al., 2019). Co-occurrence networks were obtained, with each node representing one OTU and each edge denoting a strong and significant connection. Network visualization and calculations of network topological properties (e.g., degree and modularity) were performed using the interactive Gephi platform (Xiong et al., 2021b). Cladograms for taxonomy were drawn using the R package Metacoder (Foster et al., 2017). All the statistical analyses for data were performed using the R software v3.6.0.
Results
Microbial diversity and networks in ECM roots
Bacterial and fungal communities associated to ECM roots of stone oaks and Yunnan pines were characterized by metabarcoding rDNA sequencing. In total, 4,791,454, 8,160,173 and 4,235,698 high-quality sequences were obtained and clustered into 6,081, 1,284 and 916 OTUs for 16S, ITS1 and ITS2, respectively. Lithocarpus ECM roots had a significantly higher bacterial alpha-diversity (observed OTUs and Shannon index) than Pinus ECM roots (p < 0.01, one-way ANOVA, Figure 1A). This bacterial alpha-diversity was positively correlated to the tree circumference (DBH; p < 0.001) for Lithocarpus ECM samples, whereas this was not the case for Pinus ECM roots (Supplementary Figure S2). Pine ECM roots displayed a significantly higher fungal alpha-diversity than Lithocarpus ECM roots (Figures 1B,C). No correlation between this alpha-diversity and tree circumference was observed (Supplementary Figure S2).
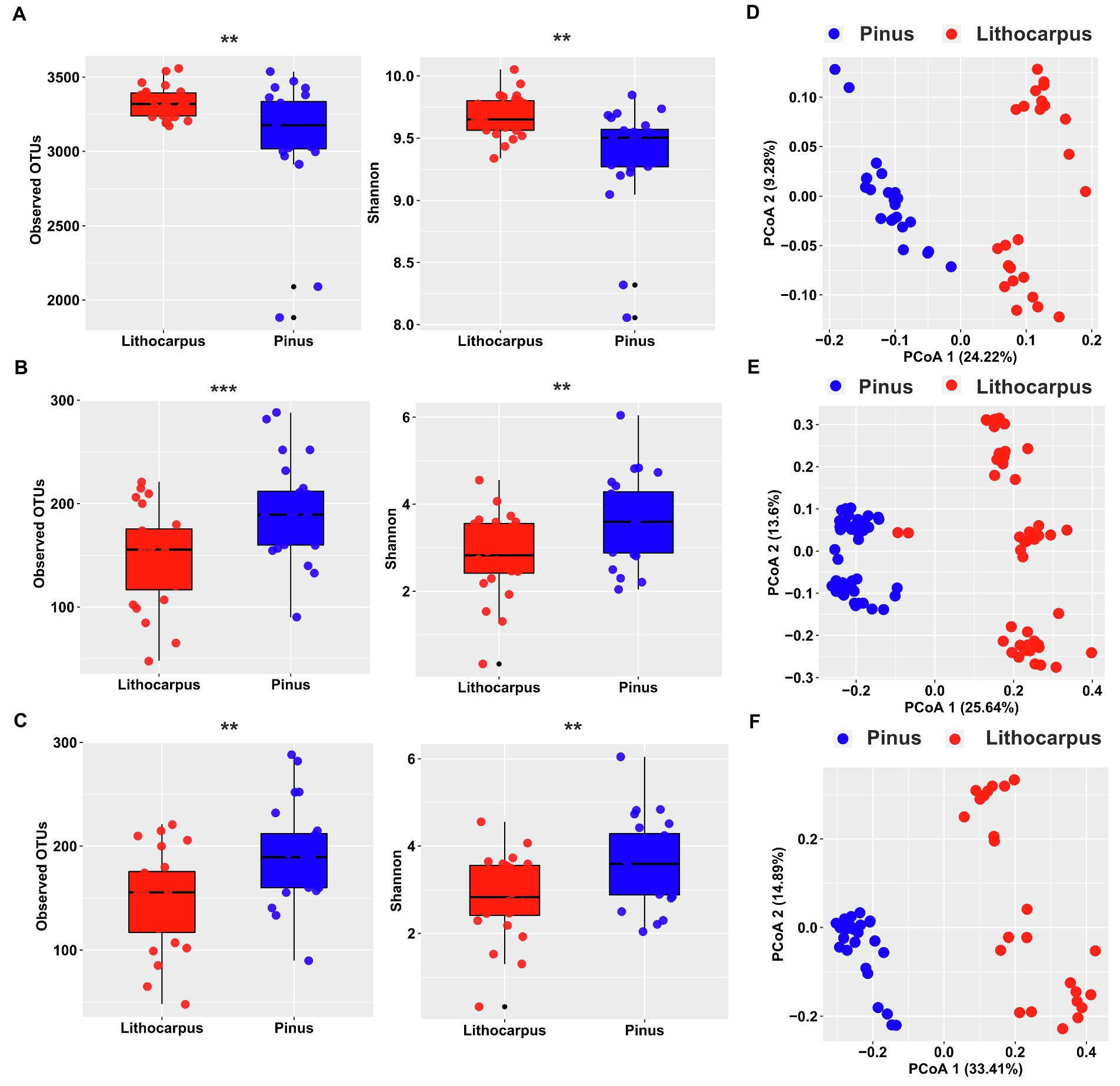
Figure 1. Diversity of bacterial and fungal communities associated to Lithocarpus and Pinus ECM roots. Alpha-diversity measurements are based on the observed OTUs and Shannon index for the bacterial (A) and fungal (B, ITS1; C, ITS2) microbiomes. Unconstrained PCoA for beta-diversity using Bray-Curtis distances in bacterial (D) and fungal (E, ITS1; F, ITS2) communities identified in Lithocarpus and Pinus ECM roots. Statistical data analyses were performed using one-way ANOVA (**p < 0.01, ***p < 0.001). For the alpha-and beta-diversity analyses based on ITS1 sequences we used samples collected during both the dry and wet seasons, while for the analyses based on ITS2 sequences we only used the samples collected during the wet season.
Both bacterial and fungal beta-diversities were mainly impacted by the host tree (Lithocarpus vs. Pinus; R2 > 22%, p = 0.001). Seasonality also exerted a significant influence on the fungal community, but only accounted for a slight variation of the fungal OTU composition (9.79%; Figures 1D–F; Supplementary Table S1). The position of the soil sampling location relative to the tree trunk did not significantly influence the root microbiome (Supplementary Table S1).
To gain a deeper insight into the interactions among microorganisms, we performed a co-occurrence network analysis for each tree species. The microbial networks obtained for bacterial and fungal communities followed similar trends. Their complexity was higher in Lithocarpus ECM roots compared to Pinus ECM roots, but their modularity was higher in Pinus ECM roots (Figure 2; Table 1). We also detected more hub nodes in the networks associated with Lithocarpus ECM roots compared to Pinus ECM roots. Bacterial hub nodes pertained to the Actinobacteria and Proteobacteria phyla, whereas fungal hub nodes pertained to the Agaricomycetes class (Figure 2; Supplementary Table S2). The bacterial network associated with Lithocarpus ECM roots was enriched in OTUs pertaining to Planctomycetes (p < 0.05, fisher’s exact test, Supplementary Table S3). Fungal networks associated with Lithocarpus ECM roots were enriched in OTUs pertaining to Agaricomycetes (based on ITS1), whereas those associated with Pinus ECM roots were enriched in Eurotiomycetes and Archaeorhizomycetes OTUs (based on both ITS1 and ITS2).
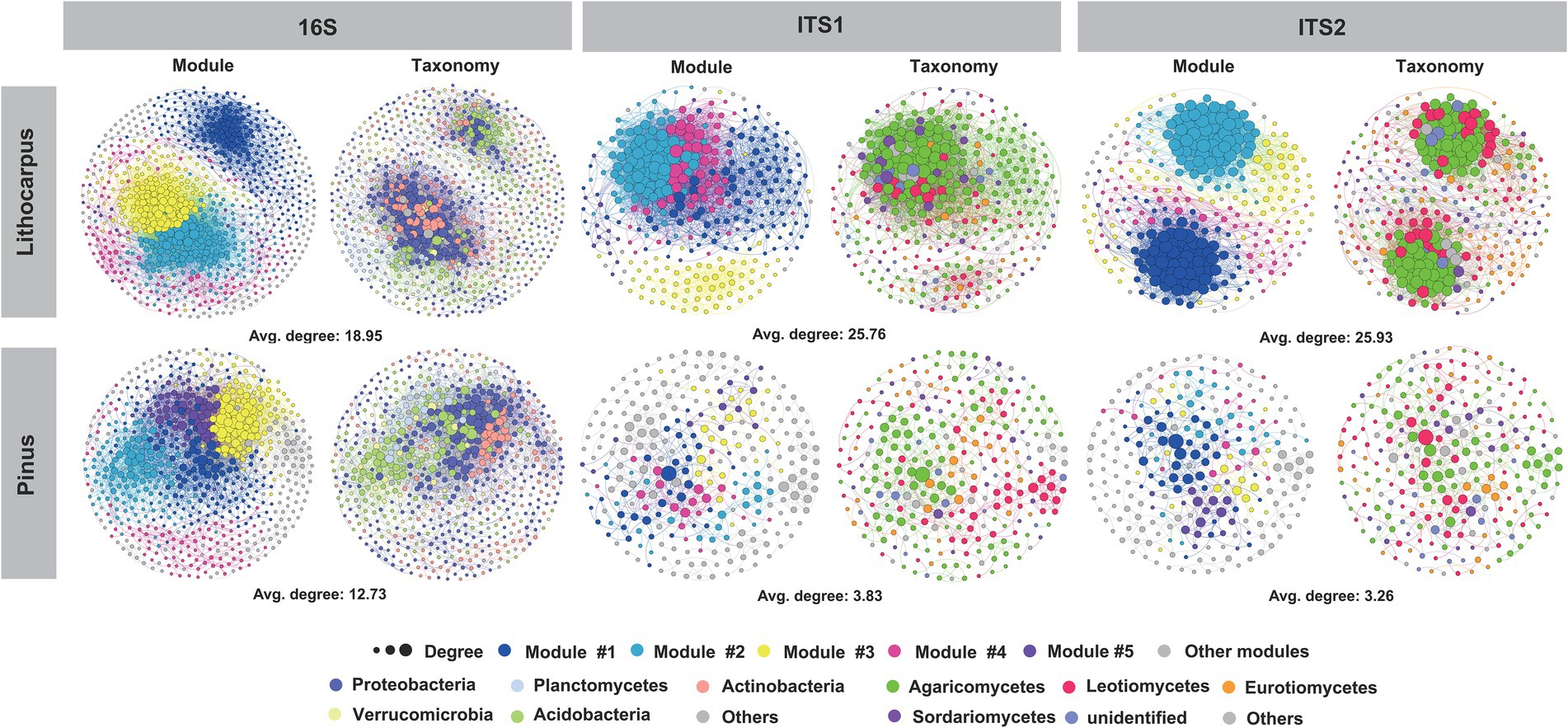
Figure 2. The co-occurrence networks of bacterial and fungal OTUs in Lithocarpus and Pinus ECM roots. The nodes in the network are colored based on phylum and class level or modularity class. The edge thickness is proportional to the weight of each correlation and node size is proportional to the degree of each OTUs. The co-occurrence network analysis based on ITS1 sequences have been carried out using samples collected during the wet season only.
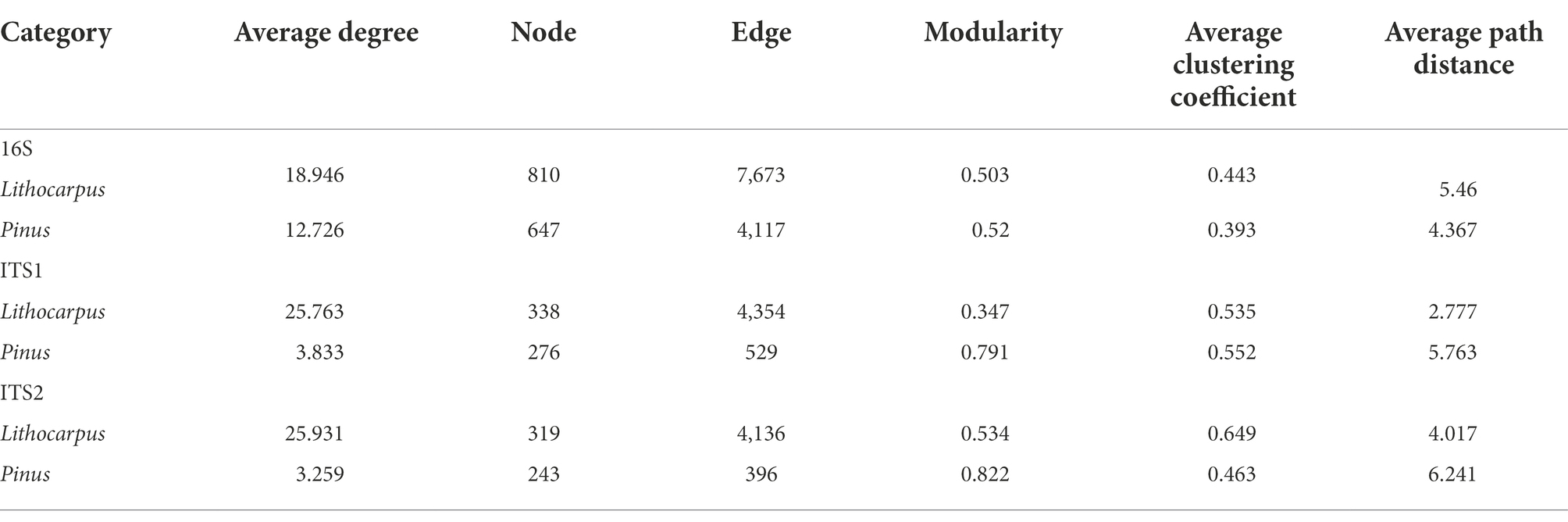
Table 1. Topological properties of the co-occurrence networks of Lithocarpus and Pinus ECM roots.
Composition of bacterial and fungal communities of ECM roots
The bacterial species identified in ECM roots belong to Proteobacteria (44.3%), Acidobacteria (20.3%), Actinobacteria (19.3%), Planctomycetes (4.5%) and Verrucomicrobia (3.7%). Rhizobiales (13.6%) and Acidobacteriales (13.2%) dominated this bacterial community leaving on the surface or inside ECM roots (Supplementary Figure S3). Pine ECM roots were enriched in Actinobacteria, Cyanobacteria, Tenericutes and Dependentiae with Actinobacteria being the most important biomarker taxa. Lithocarpus ECM roots were enriched in Acidobacteria, Bacteroidetes, Verrucomicrobia, Armatimonadetes and Planctomycetes with Acidobacteria being the most important biomarker taxa (Welch’s test, p < 0.05; Figure 3A; Supplementary Figure S4 A). Interestingly, Pinus ECM roots comprised a higher proportion of specific OTUs relative to Lithocarpus, while the latter had more significantly enriched OTUs (Figures 3B,C). OTUs annotated as Alphaproteobacteria, Actinobacteria, Thermoleophilia, Acidimicrobiia, Babeliae, Ktedonobacteria and Melainabacteria were more abundant in Pinus ECM roots, whereas Bacteroidia, Deltaproteobacteria, Verrucomicrobiae, Clostridia, Bacilli, Anaerolineae, Blastocatellia (subgroup 4) and Acidobacteria (subgroup 6) were more abundant in Lithocarpus-ECM roots (Fisher exact test, p < 0.05; Supplementary Table S4).
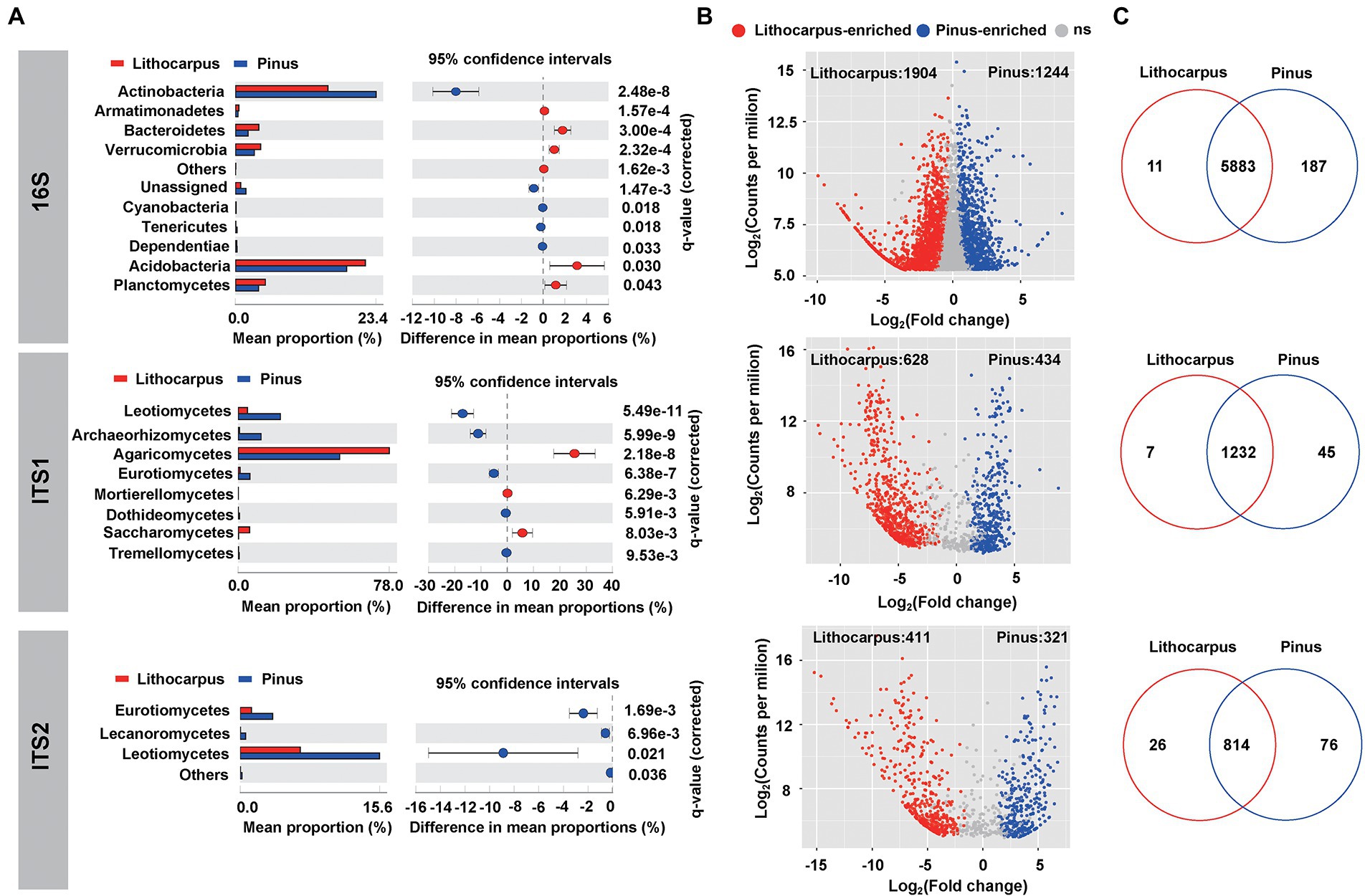
Figure 3. Tree species shape the microbial composition of ECM roots. (A) Differential abundance of bacterial and fungal OTUs in Lithocarpus and Pinus ECM roots. Welch’s tests followed by Benjamini-Hochberg FDR corrections were performed between Lithocarpus and Pinus ECM roots at phylum (bacterial OTUs) and class (fungal OTUs) levels. (B) The volcano plot shows the enriched OTUs in Lithocarpus and Pinus ECM roots. Each dot represents a single OTU. Each red dot represents an individual enriched OTUs in Lithocarpus ECM roots and each blue dot represents an individual enriched OTUs in Pinus ECM roots. The x-axis represents the fold-change in abundance and the y-axis represents the average OTUs abundance (in counts per million, CPM). (C) Venn diagrams showing the shared and specific bacterial and fungal OTUs among Lithocarpus and Pinus ECM roots. The ITS1 sequences used for this analysis were produced from samples harvested during both the dry and wet seasons.
In the stone oak old-growth forest and the pine woodlands in Ailaoshan, ECM fungi, such as Russula, Lactarius and Lactifluus in the Russulaceae, were the most abundant OTUs detected in the soil fungal community, irrespective of the soil layers and season (Zeng et al., 2022). As shown by the present analyses, the fungal community of ECM roots was significantly different between the tree species and the sampling season had also a significant influence on this root-associated mycobiome (Figure 1; Supplementary Table S1). It was dominated by Agaricomycetes with a total relative abundance of 65 to 78% based on ITS1 or ITS2 sequencing, respectively (Supplementary Figures S5, S6). The most abundant OTUs associated to Lithocarpus roots belong to ECM taxa, such as Russula, Tomentella and Laccaria, whereas Tomentella, Tylospora and Inocybe were dominant in pine roots (Supplementary Figures S5, S6). Of note, a large proportion of OTUs comprised unidentified fungal taxa. In addition, Leotiomycetes, Archaeorhizomycetes, Eurotiomycetes, Dothideomycetes, and Tremellomycetes was significantly higher in stone oak ECM roots, whereas Agaricomycetes, Saccharomycetes and Mortierellomycetes were significantly lower in pine ECM roots (Welch’s test, p < 0.05; Figure 3A). Notably, we found substantial differences in OTU distributions according to the rDNA ITS region used for metabarcoding (ITS1 vs. ITS2; Supplementary Figures S7, S8). For example, the LEfSe analysis identified Tomentella in the Thelephoraceae (Supplementary Figure S4) as the main biomarker taxa for Pinus ECM roots, while the ITS1-based survey identified Leotiomycetes and Archaeorhizomyces taxa (Supplementary Figure S4). For Lithocarpus ECM roots, Russula was the major biomarker taxa based on both ITS1-and ITS2-sequencing.
Enriched ITS1-related OTUs affiliated to Russulaceae, Sebacinaceae, Boletaceae, Cortinariaceae, Hydnangiaceae, Tricholomataceae, Leotiaceae, Cephalothecaceae and Mortierellaceae were more abundant in Lithocarpus, while those annotated as Herpotrichiellaceae, Myxotrichaceae, Aspergillaceae, Inocybaceae, and Trichocomaceae were more abundant in Pinus (Fisher’s exact test, p < 0.05; Supplementary Table S5). On the other hand, enriched ITS2-related OTUs belonging to Russulaceae, Hyaloscyphaceae, Tricholomataceae, Cortinariaceae, Hydnangiaceae, Sebacinaceae and Gomphaceae were more abundant in Lithocarpus, while those corresponding to Atheliaceae, Myxotrichaceae, Inocybaceae, Trichocomaceae and Vibrisseaceae were more abundant in Pinus (Fisher’s exact test, p < 0.05; Supplementary Table S6). This discrepancy in OTU distribution related to the ITS sequences used for metabarcoding has been reported previously (De Beeck et al., 2014; Supplementary Figure S8) and thus, use of both ITS1 and ITS2 sequences are recommended for OTU surveys.
Effect of seasonality on the fungal microbiome of ECM roots
The OTU richness and diversity of ECM roots sampled during the dry season were higher than those collected during the wet season. Notably, the observed OTU indexes for ECM roots harvested at the end of the dry season were significantly higher than those sampled at the end of the wet season (p < 0.05, one-way ANOVA; Figure 4A). Seasonality had a significant impact on beta-diversity (Supplementary Table S1). On our principal coordinate analysis (PCoA), the two main coordinates explained 39.24% of the variation, of which PC1 accounted for 25.64%, while PC2 for 13.6% of the total variation (Figure 4B).
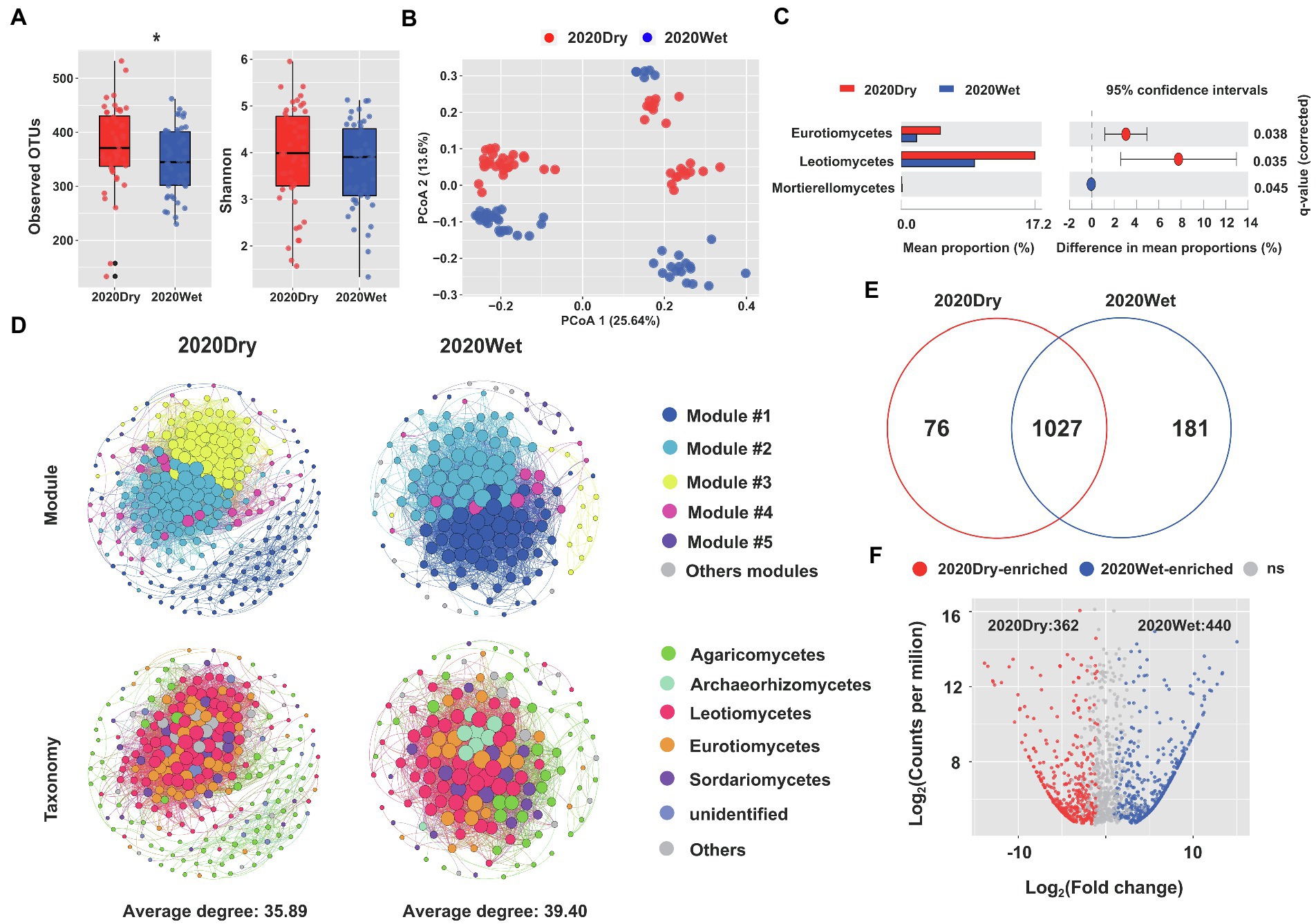
Figure 4. Seasonality influences the fungal microbiome of ECM roots. (A) Alpha-diversity measurements are based on the observed OTUs and Shannon index for the fungal OTUs. Statistical data analyses were performed using one-way ANOVA (*p < 0.05). (B) PCoA of beta-diversity using Bray-Curtis distances for fungal OTUs. (C) Differential abundances of fungal OTUs in ECM roots at the end of the dry season (2020Dry) and at the end of the wet season (2020Wet). Welch’s tests followed by Benjamini-Hochberg FDR corrections were performed for different sampling seasons. (D) Co-occurrence networks in fungal communities at the end of the dry season (2020Dry) and at the end of the wet season (2020Wet). (E) Venn diagrams showing the shared and specific fungal OTUs identified at the end of the dry season (2020Dry) and at the end of the wet season (2020Wet). (F) The Volcano plot displays the enriched OTUs for each sampling season. Each dot represents a single out, while red and blue dots represent an individual enriched OTUs identified at the end of the dry season (2020Dry) and at the end of the wet season (2020Wet), respectively.
The complexity of the microbiome networks taking place in ECM roots was higher during the wet season by comparison to the dry season, but the annotated nodes were not significantly different (Figure 4D; Supplementary Table S7). We found a higher abundance of Eurotiomycetes and Leotiomycetes and a lower abundance of Mortierellomycetes in dry season-samples compared to the wet season-samples (Welch’s test, p < 0.05; Figure 4C). The Leotiomycetes class was identified as the most significant marker of the dry season and the Tomentella genus was the most significant marker of the wet season (Supplementary Figure S9); 362 OTUs (e.g., Aspergillaceae, Trichocomaceae, Helotiaceae) and 440 OTUs (e.g., Thelephoraceae, Boletaceae, Cordycipitaceae, Amanitaceae and Gomphaceae) were significantly enriched in the ECM roots collected at the end of the dry season and wet seasons, respectively (Fisher’s exact test, p < 0.05; Figures 4E,F; Supplementary Table S8).
Discussion
Forest trees are intimately associated with hundreds of microorganisms that contribute substantially to their biology. They are supra-organisms hosting a wide range of commensal, beneficial and detrimental bacteria and fungi. The tree with its associated microbiome—the collection of all microorganisms in a location—faces altered environmental conditions as a result of forest replacement and a rapidly changing climate. Characterizing the mechanisms shaping the tree microbiome is therefore required for a better understanding of tree fitness and adaptation to changing environments, and the ecology of forest ecosystems (Hacquard, 2016; Mishra et al., 2020; Hacquard et al., 2022). As tree roots are associated to hundreds of ECM fungi, there is a need to characterize the communities of bacteria and fungi inhabiting ECM roots (Garbaye, 1994; Perotto and Bonfante, 1997; Bending et al., 2002; Frey Klett et al., 2007; Marupakula et al., 2016). Several studies have characterized the bacterial communities associated with ECM roots (Izumi et al., 2007, 2008; Uroz et al., 2010; Vik et al., 2013; Nguyen and Bruns, 2015; Marupakula et al., 2016, 2017). On the other hand, little information is available on the fungal communities, i.e., the mycobiome, associated to ECM roots (Izumi and Finlay, 2011; Marjanović et al., 2020). In the present study, we surveyed both bacterial and fungal communities of roots from stone oaks and Yunnan pines, two dominant species of the subtropical forest associations found in the subtropical Ailaoshan. Our findings confirmed and extended previous studies in showing that the ECM microbiome is mainly shaped by the host tree. In addition, we found that seasonality had a significant effect on the fungal diversity and microbial network complexity of stone oak and pine ECMs.
Distribution of bacterial and fungal OTUs in Lithocarpus and Pinus ECM roots
The diversity and composition of the bacterial microbiome of stone oak and Yunnan pine ECM roots were very similar with Actinobacteria, Acidobacteria and Proteobacteria dominating this community. Previous studies have also failed to demonstrate substantial differences between bacterial microbiomes associated with ECM roots (Burke et al., 2006; Izumi et al., 2007). In contrast, Izumi et al. (2007) have shown that the roots of B. pubescens colonized by various ECM fungi hosted distinct bacterial and ascomycetous communities. One explanation to explain the observed discrepancies between studies may be differential flow of carbohydrates and other nutrients from ECM fungi to the bacteria depending on the host tree species and age. The lack of any systematic variation in bacterial community structure related to the presence or absence of particular ECM fungal symbionts may also be related to the fact that temporal variation may confound systematic effects of the dominant mycorrhizal host fungi on bacterial community structure (Marupakula et al., 2016). The dominant orders found in Lithocarpus and Pinus ECM roots were Acidobacteriales, Rhizobiales, Betaproteobacteriales and Frankiales. Further studies will be required to elucidate the functional significance of these bacterial taxa but there is a broad correspondence between the groups found here and those identified in other studies of ECM roots. The presence of a core bacterial microbiome in ECM roots suggests that many of the bacterial genera were persistent in different ECM roots (i.e., from stone oak and pine), although there were changes in their relative abundance. This study highlights the diversity of bacteria associated with roots of major tree species growing in subtropical forests.
The fungal microbiome of stone oak and pine ECM roots was strikingly different at the OTU level, suggesting that the two tree species are recruiting a different set of ECM symbionts. As a consequence, this diversity impacts the cortege of non-symbiotic fungi inhabiting ECM roots. Russula, Tomentella, Laccaria and Sebacina taxa were the most abundant OTUs in Lithocarpus ECM roots and they are those forming the sampled ECM roots. OTUs belonging to commensal/saprotrophic fungi were also identified, including Candida, Penicillium, Leotia and Oidiodendron taxa. In Pinus ECM roots, the symbiotic Tomentella and Tylospora taxa were abundant, but a large proportion of OTUs belong to saprotrophic and endophytic taxa, such as Archaeorhizomyces and Oidiodendron. Tree phenology and thus leaf litter, but also metabolites released by roots, have a major impact on the rhizospheric microbial communities and recruitment of ECM symbionts (Frey Klett et al., 2007; Boberg et al., 2014), likely explaining the differences observed here in the distribution of ECM symbionts. In addition to root exudates, hyphal exudates, such as trehalose, released by the various ECM fungi played an important role in recruiting bacterial and fungal microbiomes (Zhang et al., 2021). We have shown in our companion study (Zeng et al., 2022) that the edaphic features, such as soil pH and nutrient content, were significantly different in the Lithocarpus old-growth forest by comparison to the Pinus woodland. These differences in edaphic features influenced the soil microbial communities (Zeng et al., 2022), but also the ECM microbial communities (present study) as shown in other forest ecosystems (Peay et al., 2013; Liu et al., 2014, 2015; He et al., 2017). Given that soil microbial communities feed the plant microbiome, both the reservoir of microorganisms in soil adjacent to roots and the soil edaphic parameters strongly influence the plant microbiome (Frey Klett et al., 2007; Bonito et al., 2014).
Our results showed that the alpha-diversity of the bacterial community from Lithocarpus ECM roots was significantly higher than that of Pinus ECM roots, while the trend was reversed for the fungal microbiome. Moreover, the microbial network occurring in Lithocarpus ECM roots was higher than that of Pinus ECM roots with a significantly different OTU composition (Figures 1, 2). A positive relationship between the alpha-diversity and tree circumference of Lithocarpus for the bacterial community was observed (Supplementary Figure S2). Of note, it has been reported that the ECM alpha diversity is not strongly linked to tree growth, while variations in the beta diversity of ECM community is the strongest predictor of tree growth rate across Europe (Anthony et al., 2022).
Seasonality influences the ECM root mycobiome
The Ailaoshan resides in the center of the largest subtropical land area in the world (Young and Wang, 1989). This region is a major climatic border between China southwestern and southeastern monsoon systems, and the northern Himalaya Plateau. Influenced by the southwest and southeast monsoons, the climate of the Ailaoshan alternates between wet and dry conditions. Mean annual precipitation is 1799 mm, 86% of which occurs during the monsoon rainy season from May to October (Song et al., 2017). Therefore, seasonal variability in soil water content is substantial (Wu et al., 2014) and influences the soil microbial communities (Zeng et al., 2022). As soil water content affects the soil mycobiome composition (Zeng et al., 2022) and competition between ECM and saprotrophic fungi (Baldrian, 2017), we surveyed the ECM root mycobiome at the end of the wet or dry seasons. The alpha-diversity of the fungal community of ECM roots at the end of the dry season was higher than at the end of the wet season. Furthermore, samples collected during the dry season showed a higher abundance of saprotrophic Eurotiomycetes and Leotiomycetes, while ECM roots collected during the wet season were enriched in saprotrophic Mortierellomycetes. The observed seasonal changes in the ECM root mycobiome may be linked to variation in root metabolic activities. Frequent soil droughts taking place during the dry season may also favor ECM root senescence and decay, providing favorable conditions for the establishment of saprotrophic fungi.
Fungal identification biases
As reported in previous studies, the recovered taxonomic richness of the fungal community differs among selected barcode-primer pair combinations for amplifying the rDNA ITS (De Beeck et al., 2014; Tedersoo et al., 2015; Mbareche et al., 2020). ITS1 and ITS2 sequencing provided consistent results in ranking taxonomic richness and recovering the importance of tree species in driving fungal community composition in stone oak and pine ECM roots, except for the Archaeorhizomycetes and Tremellomycetes which were mainly detected by ITS1 sequencing and Lecanoromycetes was more abundant in ITS2 sequencing (Supplementary Figures S7, S8).
Conclusion
In conclusion, the present study provides new information about the identity and diversity of different bacterial and fungal microbiomes associated with different types of ECM associations from two major native tree species of a subtropical mountain forests. As reported previously, our results confirmed the major influence of the tree species on the composition of the ECM mycobiome. On the other hand, the bacterial communities associated with ECM roots were less influenced by the changes in the host tree and associated ECM fungal communities. This work paves the way for more detailed studies of the function expressed by the communities of ECM fungi and their associated communities of bacteria and fungi. A better understanding of the interactions between bacteria and fungi in the mycorrhizosphere of stone oaks and Yunnan pines will be critical to understand the soil–plant interface in the threatened subtropical forests.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
QZ and XM collected the samples and extracted DNA. QZ analyzed the data with help from AL and XM. FM and YD coordinated the project and designed the experimental plan. QZ and FM wrote the manuscript with input from AL. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Beijing Advanced Innovation Center for Tree Breeding by Molecular Design and the Laboratory of Excellence ARBRE (grant number ANR-11-LABX-0002-01). We also acknowledge grants from the China Postdoctoral Science Foundation (2019M660508 to QZ), the National Natural Science Foundation of China (grant number U1802231 to YD), and the Second Tibetan Plateau Scientific Expedition and Research Program (STEP, No. 2019QZKK0503 to YD).
Acknowledgments
We would like to acknowledge the support of Haixia Wu and Xinhao Li from the Research Institute of Resource Insects, Chinese Academy of Forestry, and Zhiyun Lu, Associate Director of the Ailaoshan Subtropical Forest Ecosystem Research Station.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.916337/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Different morphotypes of ECM roots sampled beneath Lithocarpus and Pinus trees in the Ailaoshan old-growth forest and pine woodland and the protocol used for the DNA metabarcoding survey.
SUPPLEMENTARY FIGURE S2 | Linear regression showing the relationship between the tree circumference (BDH) and alpha-diversity of bacterial and fungal OTUs (observed OTUs and Shannon index).
SUPPLEMENTARY FIGURE S3 | Relative abundance of the most abundant bacterial OTUs in Lithocarpus and Pinus ECM roots at phylum and order levels. The average relative abundance phyla or class with less than 0.05% across all samples are grouped into “Others”. (A) At phylum level, (B) At order level.
SUPPLEMENTARY FIGURE S4 | The biomarker taxa of bacterial and fungal communities varied among Lithocarpus and Pinus ECM roots. LEfSe identified the biomarker taxa associated with Lithocarpus and Pinus ECM roots, respectively. Only the top 10 most specific biomarker taxa are displayed. (A) 16S rDNA sequencing, (B) ITS1 sequencing, (C) ITS2 sequencing.
SUPPLEMENTARY FIGURE S5 | Relative abundance of the most abundant fungal OTUs in Lithocarpus and Pinus ECM roots at class, order and family levels based on ITS1 sequencing. The average relative abundance order or family with less than 0.05% across all samples are grouped into “Others”. (A) At class level, (B) At family level, (C) At genus level.
SUPPLEMENTARY FIGURE S6 | Relative abundance of the most abundant fungal OTUs in Lithocarpus and Pinus ECM roots at class, order and family levels based on ITS2 sequencing. The average relative abundance order or family with less than 0.05% across all samples are grouped into “Others”. (A) At class level, (B) At family level, (C) At genus level.
SUPPLEMENTARY FIGURE S7 | The choice of ITS region impacts the identification of fungal OTUs of ECM roots. (A) Relative abundances of the enriched fungal taxa using ITS1 and ITS2 primers at the class level. The average relative abundant classes less than 0.05% across all samples are grouped into “Others”. (B) Differential abundances of fungal OTUs identified by using ITS1 or ITS2 sequences. Welch’s tests followed by Benjamini-Hochberg FDR corrections were performed among different ITS. For these analyses, we used samples collected during the wet seasons.
SUPPLEMENTARY FIGURE S8 | Taxonomic distribution of the most abundant fungal OTUs identified using either ITS1 or ITS2 sequences. Blue and red circles represent the microbial taxonomic ranks with are found as significantly more abundant based on ITS1 or ITS2 sequences, respectively. The color of nodes represents the value of the Log2 ratio of ITS1 and ITS2 with OTU count (Wilcoxon Rank Sum test, P<0.05). Only the OTUs with an average relative abundance >0.1% are displayed. For these analyses, we used samples collected during the wet seasons.
SUPPLEMENTARY FIGURE S9 | The biomarker taxa identified in ECM roots at the end of the dry season (2020Dry) and at the end of the wet season (2020Wet) based on ITS1 sequencing. Only the top 10 most specific biomarker taxa are displayed.
SUPPLEMENTARY TABLE S1 | Effects of multiple biotic and abiotic factors on the root microbiome composition of Lithocarpus and Pinus ECM roots.
SUPPLEMENTARY TABLE S2 | Comparison of nodes assignments between Lithocarpus and Pinus ECM roots.
SUPPLEMENTARY TABLE S3 | The hubs of microbial co-occurrence networks of Lithocarpus and Pinus ECM roots.
SUPPLEMENTARY TABLE S4 | Comparison of enriched bacterial OTUs between Lithocarpus and Pinus ECM roots based on 16S sequencing.
SUPPLEMENTARY TABLE S5 | Comparison of enriched fungal OTUs between Lithocarpus and Pinus ECM roots based on ITS1 sequencing.
SUPPLEMENTARY TABLE S6 | Comparison of enriched fungal OTUs between Lithocarpus and Pinus ECM roots based on ITS2 sequencing.
SUPPLEMENTARY TABLE S7 | Comparison of node assignments between ECM roots sampled at the end of the dry season (2020Dry) or at the end of the wet season (2020Wet) based on ITS2 sequencing.
SUPPLEMENTARY TABLE S8 | Comparison of node assignments between ECM roots sampled at the end of the dry season (2020Dry) or at the end of the wet season (2020Wet) based on ITS2 sequencing.
References
Andrade, G., Mihara, K. L., Linderman, R. G., and Bethlenfalvay, G. J. (1997). Bacteria from rhizosphere and hyphosphere soils of different arbuscular-mycorrhizal fungi. Plant Soil 192, 71–79. doi: 10.1023/A:1004249629643
Anthony, M. A., Crowther, T. W., van der Linde, S., Suz, L. M., Bidartondo, M. I., Cox, F., et al. (2022). Forest tree growth is linked to mycorrhizal fungal composition and function across Europe. ISME J. 16, 1327–1336. doi: 10.1038/s41396-021-01159-7
Aspray, T. J., Jones, E. E., Davies, M. W., Shipman, M., and Bending, G. D. (2013). Increased hyphal branching and growth of ectomycorrhizal fungus Lactarius rufus by the helper bacterium Paenibacillus sp. Mycorrhiza 23, 403–410. doi: 10.1007/s00572-013-0483-1
Baldrian, P. (2017). Forest microbiome: diversity, complexity and dynamics. FEMS Microbiol. Rev. 41, 109–130. doi: 10.1093/femsre/fuw040
Barberan, A., Bates, S. T., Casamayor, E. O., and Fierer, N. (2014). Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 6:343. doi: 10.1038/ismej.2013.236
Beckers, B., De Beeck, M. O., Weyens, N., Boerjan, W., and Vangronsveld, J. (2017). Structural variability and niche differentiation in the rhizosphere and endosphere bacterial microbiome of field-grown poplar trees. MICROBIOME. 5:25. doi: 10.1186/s40168-017-0241-2
Bending, G. D., Poole, E. J., Whipps, J. M., and Read, D. J. (2002). Characterisation of bacteria from Pinus sylvestris–Suillus luteusmycorrhizas and their effects on root–fungus interactions and plant growth. FEMS Microbiol. Ecology 39, 219–227. doi: 10.1016/S0168-6496(01)00215-X
Berendsen, R. L., Pieterse, C. M. J., and Bakker, P. A. H. M. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Berg, G., Grube, M., Schloter, M., and Smalla, K. (2014). Unraveling the plant microbiome: looking back and future perspectives. Front. Microbiol. 5:148. doi: 10.3389/fmicb.2014.00148
Boberg, J. B., Finlay, R. D., Stenlid, J., Ekblad, A., and Lindahl, B. D. (2014). Nitrogen and carbon reallocation in fungal mycelia during decomposition of boreal forest litter. PLoS One 9:e92897. doi: 10.1371/journal.pone.0092897
Bonito, G., Reynolds, H., Robeson, M. S., Nelson, J., Hodkinson, B. P., Tuskan, G., et al. (2014). Plant host and soil origin influence fungal and bacterial assemblages in the roots of woody plants. Mol. Ecol. 23, 3356–3370. doi: 10.1111/mec.12821
Burke, D. J., Kretzer, A. M., Rygiewicz, P. T., and Topa, M. A. (2006). Soil bacterial diversity in a loblolly pine plantation: influence of ectomycorrhizas and fertilization. FEMS Microbiol. Ecol. 57, 409–419.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. P. Natl. Acad. Sci. Usa. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Chan, O. C., Yang, X., Fu, Y., Feng, Z., Sha, L., Casper, P., et al. (2006). 16S rRNA gene analyses of bacterial community structures in the soils of evergreen broad-leaved forests in south-West China. FEMS Microbiol. Ecol. 58, 247–259. doi: 10.1111/j.1574-6941.2006.00156.x
De Beeck, M. O., Lievens, B., Busschaert, P., Declerck, S., Vangronsveld, J., and Colpaert, J. V. (2014). Comparison and validation of some its primer pairs useful for fungal metabarcoding studies. PLoS One 9:7629. doi: 10.1371/journal.pone.0097629
Deveau, A. (2016). How does the tree root microbiome assemble? Influence of ectomycorrhizal species on Pinus sylvestris root bacterial communities. Environ. Microbiol. 18, 1303–1305. doi: 10.1111/1462-2920.13214
Dreischhoff, S., Das, I. S., Jakobi, M., Kasper, K., and Polle, A. (2020). Local responses and systemic induced resistance mediated by ectomycorrhizal fungi. Front. Plant Sci. 11:590063. doi: 10.3389/fpls.2020.590063
Emmett, B. D., Levesque-Tremblay, V., and Harrison, M. J. (2021). Conserved and reproducible bacterial communities associate with extraradical hyphae of arbuscular mycorrhizal fungi. ISME J. 15, 2276–2288. doi: 10.1038/s41396-021-00920-2
Foster, Z. S. L., Sharpton, T. J., and Grunwald, N. J. (2017). Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol. 13:e1005404. doi: 10.1371/journal.pcbi.1005404
Frey Klett, P., Garbaye, J., and Tarkka, M. (2007). The mycorrhiza helper bacteria revisited. New Phytol. 176, 22–36. doi: 10.1111/j.1469-8137.2007.02191.x
Gahan, J., and Schmalenberger, A. (2015). Arbuscular mycorrhizal hyphae in grassland select for a diverse and abundant hyphospheric bacterial community involved in sulfonate desulfurization. Appl. Soil Ecol. 89, 113–121. doi: 10.1016/j.apsoil.2014.12.008
Garbaye, J. (1994). Tansley review no. 76. Helper bacteria: A new dimension to the mycorrhizal symbiosis. New Phytol. 128, 197–210. doi: 10.1111/j.1469-8137.1994.tb04003.x
Hacquard, S. (2016). Disentangling the factors shaping microbiota composition across the plant holobiont. New Phytol. 209, 454–457. doi: 10.1111/nph.13760
Hacquard, S., Wang, E., Slater, H., and Martin, F. (2022). Impact of global change on the plant microbiome. New Phytol. 234, 1907–1909.
He, J., Tedersoo, L., Hu, A., Han, C., He, D., Wei, H., et al. (2017). Greater diversity of soil fungal communities and distinguishable seasonal variation in temperate deciduous forests compared with subtropical evergreen forests of eastern China. FEMS Microbiol. Ecol. 93:069. doi: 10.1093/femsec/fix069
Izumi, H., Cairney, J. W., Killham, K., Moore, E., Alexander, I. J., and Anderson, I. C. (2008). Bacteria associated with ectomycorrhizas of slash pine (Pinus elliottii) in South-Eastern Queensland, Australia. FEMS Microbiol. Lett. 282, 196–204. doi: 10.1111/j.1574-6968.2008.01122.x
Izumi, H., and Finlay, R. D. (2011). Ectomycorrhizal roots select distinctive bacterial and ascomycete communities in Swedish subarctic forests. Environ. Microbiol. 13, 819–830. doi: 10.1111/j.1462-2920.2010.02393.x
Izumi, H., Moore, E. R. B., Killham, K., Alexander, I. J., and Anderson, I. C. (2007). Characterisation of endobacterial communities in ectomycorrhizas by DNA-and RNA-based molecular methods. Soil Biol. Biochem. 39, 891–899. doi: 10.1016/j.soilbio.2006.10.011
Liu, J., Sui, Y., Yu, Z., Shi, Y., Chu, H., Jin, J., et al. (2014). High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of Northeast China. Soil Biol. Biochem. 70, 113–122. doi: 10.1016/j.soilbio.2013.12.014
Liu, J., Sui, Y., Yu, Z., Shi, Y., Chu, H., Jin, J., et al. (2015). Soil carbon content drives the biogeographical distribution of fungal communities in the black soil zone of Northeast China. Soil Biol. Biochem. 83, 29–39. doi: 10.1016/j.soilbio.2015.01.009
Marjanović, Ž., Nawaz, A., Stevanović, K., Saljnikov, E., Maček, I., Oehl, F., et al. (2020). Root-associated mycobiome differentiate between habitats supporting production of different truffle species in serbian riparian forests. Microorganisms. 8:1331. doi: 10.3390/microorganisms8091331
Maropola, M. K. A., Ramond, J., and Trindade, M. (2015). Impact of metagenomic DNA extraction procedures on the identifiable endophytic bacterial diversity in Sorghum bicolor (L. Moench). J. Microbiol. Methods 112, 104–117. doi: 10.1016/j.mimet.2015.03.012
Marupakula, S., Mahmood, S., and Finlay, R. D. (2016). Analysis of single root tip microbiomes suggests that distinctive bacterial communities are selected by Pinus sylvestris roots colonized by different ectomycorrhizal fungi. Environ. Microbiol. 18, 1470–1483. doi: 10.1111/1462-2920.13102
Marupakula, S., Mahmood, S., Jernberg, J., Nallanchakravarthula, S., Fahad, Z. A., and Finlay, R. D. (2017). Bacterial microbiomes of individual ectomycorrhizal Pinus sylvestris roots are shaped by soil horizon and differentially sensitive to nitrogen addition. Environ. Microbiol. 19, 4736–4753.
Mbareche, H., Veillette, M., Bilodeau, G., and Duchaine, C. (2020). Comparison of the performance of ITS1 and ITS2 as barcodes in amplicon-based sequencing of bioaerosols. PEERJ. 8:e8523. doi: 10.7717/peerj.8523
Mishra, S., Hattenschwiler, S., and Yang, X. (2020). The plant microbiome: A missing link for the understanding of community dynamics and multifunctionality in forest ecosystems. Appl. Soil Ecol. 145:103345. doi: 10.1016/j.apsoil.2019.08.007
Nazir, R., Tazetdinova, D. I., and van Elsas, J. D. (2014). Burkholderia terrae BS001 migrates proficiently with diverse fungal hosts through soil and provides protection from antifungal agents. Front. Microbiol. 5:598. doi: 10.3389/fmicb.2014.00598
Nazir, R., Warmink, J. A., Boersma, H., and van Elsas, J. D. (2010). Mechanisms that promote bacterial fitness in fungal-affected soil microhabitats. FEMS Microbiol. Ecol. 71, 169–185. doi: 10.1111/j.1574-6941.2009.00807.x
Nehls, U., and Plassard, C. (2018). Nitrogen and phosphate metabolism in ectomycorrhizas. New Phytol. 220, 1047–1058. doi: 10.1111/nph.15257
Nguyen, N. H., and Bruns, T. D. (2015). The microbiome of pinus muricata ectomycorrhizae: community assemblages, fungal species effects, and burkholderia as important bacteria in multipartnered symbioses. Microb. Ecol. 69, 914–921. doi: 10.1007/s00248-015-0574-y
Nilsson, R. H., Larsson, K., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., et al. (2019). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264. doi: 10.1093/nar/gky1022
Pan, Y., Birdsey, R. A., Fang, J., Houghton, R., Kauppi, P. E., Kurz, W. A., et al. (2011). A large and persistent carbon sink in the world's forests. Science 333, 988–993. doi: 10.1126/science.1201609
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Peay, K. G., Baraloto, C., and Fine, P. V. A. (2013). Strong coupling of plant and fungal community structure across western Amazonian rainforests. ISME J. 7, 1852–1861. doi: 10.1038/ismej.2013.66
Perotto, S., and Bonfante, P. (1997). Bacterial associations with mycorrhizal fungi: close and distant friends in the rhizosphere. Trends Microbiol. 5, 496–501. doi: 10.1016/S0966-842X(97)01154-2
Pohjanmies, T., Trivino, M., Le Tortorec, E., Mazziotta, A., Snall, T., and Monkkonen, M. (2017). Impacts of forestry on boreal forests: An ecosystem services perspective. Ambio 46, 743–755. doi: 10.1007/s13280-017-0919-5
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Riedlinger, J., Schrey, S. D., Tarkka, M. T., Hampp, R., Kapur, M., and Fiedler, H. P. (2006). Auxofuran, a novel metabolite that stimulates the growth of fly agaric, is produced by the mycorrhiza helper bacterium Streptomyces strain AcH 505. Appl. Environ. Microbiol. 72, 3550–3557. doi: 10.1128/AEM.72.5.3550-3557.2006
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). Edge R: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Song, Q., Fei, X., Zhang, Y., Sha, L., Liu, Y., Zhou, W., et al. (2017). Water use efficiency in a primary subtropical evergreen forest in Southwest China. Sci. Rep.-UK. 7:3031. doi: 10.1038/srep43031
Tedersoo, L., Bahram, M., Polme, S., Anslan, S., Riit, T., Koljalg, U., et al. (2015). Response to comment on "global diversity and geography of soil fungi". Science 349, 936. doi: 10.1126/science.aaa5594
Turner, T. R., James, E. K., and Poole, P. S. (2013). The plant microbiome. Genome Biol. 14:209. doi: 10.1186/gb-2013-14-6-209
Uroz, S., Buée, M., Murat, C., Frey-Klett, P., and Martin, F. (2010). Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Environ. Microbiol. Rep. 2, 281–288. doi: 10.1111/j.1758-2229.2009.00117.x
van der Heijden, M. G. A., Martin, F. M., Selosse, M., and Sanders, I. R. (2015). Mycorrhizal ecology and evolution: The past, the present, and the future. New Phytol. 205, 1406–1423. doi: 10.1111/nph.13288
Vik, U., Logares, R., Blaalid, R., Halvorsen, R., Carlsen, T., Bakke, I., et al. (2013). Different bacterial communities in ectomycorrhizae and surrounding soil. Sci. Rep.-UK. 3:3471. doi: 10.1038/srep03471
Wang, F., Shi, N., Jiang, R., Zhang, F., and Feng, G. (2016). In situ stable isotope probing of phosphate-solubilizing bacteria in the hyphosphere. J. Exp. Bot. 67, 1689–1701. doi: 10.1093/jxb/erv561
Warmink, J. A., Nazir, R., Corten, B., and van Elsas, J. D. (2011). Hitchhikers on the fungal highway: The helper effect for bacterial migration via fungal hyphae. Soil Biol. Biochem. 43, 760–765. doi: 10.1016/j.soilbio.2010.12.00
Wen, H. D., Lin, L. X., Yang, J., Hu, Y. H., Cao, M., Liu, Y. H., et al. (2017). Species composition and community structure of a 20 hm2 plot of mid-mountain moist ever-green broad-leaved forest on the Mts. Ailaoshan, Yunnan Province, China. Chin. J. Plant Ecol. 42, 419–429. doi: 10.17521/cjpe.2017.0272
Wu, C., Zhang, Y., Xu, X., Sha, L., You, G., Liu, Y., et al. (2014). Influence of interactions between litter decomposition and rhizosphere activity on soil respiration and on the temperature sensitivity in a subtropical montane forest in SW China. Plant Soil 381, 215–224.
Xiong, C., Singh, B. K., He, J., Han, Y., Li, P., Wan, L., et al. (2021a). Plant developmental stage drives the differentiation in ecological role of the maize microbiome. MICROBIOME. 9:171. doi: 10.1186/s40168-021-01118-6
Xiong, C., Zhu, Y., Wang, J., Singh, B., Han, L., Shen, J., et al. (2021b). Host selection shapes crop microbiome assembly and network complexity. New Phytol. 229, 1091–1104. doi: 10.1111/nph.16890
Young, S. S., Carpenter, C., and Wang, Z.-J. (1992). A study of the structure and composition of an old-growth and secondary broad-leaved forest in the Ailao Mountains of Yunnan, China. Mt. Res. Dev. 12, 269–284. doi: 10.2307/3673670
Young, S. S., and Wang, Z. J. (1989). Comparison of secondary and primary forests in the Ailao Shan region of Yunnan, China. For. Ecol. Manag. 28, 281–300. doi: 10.1016/0378-1127(89)90008-x
Zeng, Q. C., Lebreton, A., Man, X. W., Jia, L. K., Wang, G. S., Gong, S., et al. (2022). Ecological drivers of the soil microbial diversity and composition in primary old-growth forest and secondary woodland in a subtropical evergreen broad-leaved forest biome in the Ailao Mountains, China. Front. Microbiol. 13:908257. doi: 10.3389/fmicb.2022.908257
Zgadzaj, R., Garrido-Oter, R., Jensen, D. B., Koprivova, A., Schulze-Lefert, P., and Radutoiu, S. (2016). Root nodule symbiosis in Lotus japonicus drives the establishment of distinctive rhizosphere, root, and nodule bacterial communities. P. Natl. Acad. Sci. Usa. 113, E7996–E8005. doi: 10.1073/pnas.1616564113
Zhang, L., Zhou, J., George, T. S., Limpens, E., and Feng, G. (2021). Arbuscular mycorrhizal fungi conducting the hyphosphere bacterial orchestra. Trends Plant Sci. 27, 402–411. doi: 10.1016/j.tplants.2021.10.008
Keywords: mycorrhizal symbioses, community structure, evergreen forest, season, host impact
Citation: Zeng Q, Man X, Lebreton A, Dai Y and Martin FM (2022) The bacterial and fungal microbiomes of ectomycorrhizal roots from stone oaks and Yunnan pines in the subtropical forests of the Ailao Mountains of Yunnan. Front. Microbiol. 13:916337. doi: 10.3389/fmicb.2022.916337
Edited by:
Nakarin Suwannarach, Chiang Mai University, ThailandReviewed by:
Amit Kishore Singh, Tilka Manjhi Bhagalpur University, IndiaChristina Hazard, Ecole Centrale de Lyon, France
Copyright © 2022 Zeng, Man, Lebreton, Dai and Martin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yucheng Dai, yuchengdai@bjfu.edu.cn; Francis M. Martin, francis.martin@inrae.fr
†These authors have contributed equally to this work